
Abstract
Background:
Understanding how the gut microbiome adapts on exposure to individual antibiotics, with respect to antimicrobial resistance gene (ARG) enrichment, is important.
Objectives:
To characterise the changes that occur in the gut microbiome of patients enrolled in an antibiotic clinical trial and to propose methods in which to embed gut microbiome analysis into clinical trials.
Design:
This was a prospective cohort study of hospitalised patients who were successfully enrolled and randomised into two clinical trials between January 2021 to December 2021.
Methods:
Adult patients admitted to the hospital with a bloodstream infection have been randomised to receive either benzylpenicillin, ampicillin, cefazolin, ceftriaxone, piperacillin-tazobactam or meropenem at a single institution. Faecal specimens were collected at enrolment and every second day until discharge. Each specimen underwent DNA sequencing to determine microbial diversity and ARG abundance.
Results:
Ten patients (including six females) were included. DNA concentration and sampling quality were markedly lower for rectal swabs compared to stool samples. Relative abundance of Enterococcus faecium was increased in individual patients where treatment included ampicillin, meropenem and piperacillin-tazobactam. Piperacillin-tazobactam also increased the abundance of key beta-lactamase genes (blaSHV-100, blaOXA-392, blaCMY-18). Ampicillin increased the abundance of blaTEM-1A. There were no extended-spectrum beta-lactamase (ESBL) or carbapenemase genes detected in our study. The presence of key anaerobes such as Clostridium and Bifidobacterium species appeared to play an important role in colonisation resistance of E. faecium and Clostridioides difficile.
Conclusion:
Differential changes in anaerobic bacterial genera on exposure to antibiotics may be a key determinant of colonisation resistance. The pre-analytical phase of microbiome analysis is a critical factor in data quality and interpretation.
Introduction
The global burden of antimicrobial resistance continues to rise and is largely driven by antimicrobial use. 1 There exists a dire need for new management strategies to mitigate this growing risk to patient and public health outcomes. Despite being crucial for the treatment of severe infections, antimicrobial use leads to unintended consequences on the human microbiome. 2 Indeed, inappropriate and excessive use of antimicrobials also contributes to antimicrobial resistance. Within the gut microbiome, administration of broad-spectrum antimicrobials can cause a rapid and significant drop in taxonomic richness, diversity and evenness.2,3 Moreover, these compositional changes can remain long after cessation of antimicrobials. 4 In addition to changes in diversity, an increase in the abundance of antibiotic-resistance genes belonging to the resistome is observed. 5 This includes problematic antimicrobial resistance determinants such as those encoding extended-spectrum beta-lactamases (ESBL) and carbapenemases in Gram-negative bacteria, as well as vancomycin resistance genes (vanA and vanB) in Enterococcus spp. Colonisation resistance is the reverse process whereby the microbiota inhibits the expansion of Enterobacterales and enterococci in the gut lumen and is promoted by taxonomic diversity (including the presence of anaerobic bacteria). 6
It remains unclear what effect each antimicrobial has on the gut microbiome of individual patients with respect to bacterial community profiling, taxonomic diversity and antimicrobial resistance gene (ARG) abundance. Difficulties exist in carrying out such in vivo studies with no agreed-upon methodology (including statistical analysis) for the assessment and comparison of clinical specimens undergoing metagenomic sequencing. 7 Significant differences in richness and diversity have been observed within and between healthy patient stool specimens. Gaining a more refined understanding of the effect of the individual antibiotic over time, on the type and degree of change in the gut microbiome, will better inform decision making on antimicrobial prescribing. Moreover, developing an agreed-upon framework in which to embed gut microbiome analysis into contemporary clinical trials will help to compare clinically relevant antibiotic-related adverse events. 8 Here, we aim to characterise the changes that occur in the gut microbiome of patients enrolled in an interventional clinical trial receiving a single antibiotic, in addition to proposing a methods structure in which to embed gut microbiome analysis into clinical trials.
Methods
Clinical data
Adult patients admitted to the hospital with bloodstream infection at the Royal Brisbane & Women’s Hospital who had been enrolled and randomised into an interventional antibiotic clinical trial were identified prospectively for inclusion in the study between January 2021 to December 2021. Two such trials were enrolled during the study period, GAMECHANGER (ClinicalTrials.gov NCT03869437) and BALANCE (Clinical Trials.gov NCT03005145). All patients enrolled in either of these trials at our centre were eligible for inclusion in our study. Stool samples or rectal swabs were collected at baseline and every 2–3 days thereafter until discharge from the hospital. Collection of stool samples was given preference over rectal swabs by the study team. Patient demographic, microbiological and clinical data were collected prospectively. This study was approved by the Royal Brisbane & Women’s Hospital Human Research Ethics Committee (LNR/2020/QRBW/65029). Written informed consent for inclusion in our study was obtained from participants.
Sample handling and storage
Fresh faeces specimens were collected in a non-sterile container, and rectal swabs were collected in a commercial system (MWE DRYSWAB™). Faeces specimens were visually inspected, and their appearance was recorded (e.g., watery). Rectal swabs were visually inspected for discolouration immediately post-sampling (re-collection was performed if no discolouration was evident). Samples were stored at 4°C for <72 h before being transported to our reference laboratory and ultimately stored at −80°C prior to DNA extraction.
Sequencing methods
DNA extraction was performed using the QIAamp Power Fecal Pro DNA kit (Qiagen, Clayton, Victoria). Briefly, each sample was mixed with lysis buffer and sterile zirconia beads as per the manufacturer’s instructions. After bead-beating, using a modified Vortex Genie vortex mixer for 10 min (faeces) or 30 min (swabs) at room temperature, the samples were centrifuged for 5 min at 13,200 rpm and the supernatant was further treated as per the manufacturer’s recommendations. A blank control was included with each extraction run and subsequently sequenced for quality control and monitoring of possible contamination. Library preparation and sequencing were performed as previously described by Schlebusch et al. 9
Bioinformatic analysis
Human reads were quantified and removed by mapping trimmed reads to the human genome using bowtie2. 10 The raw reads were trimmed which were then used as input to MGmapper. 11 An acquired AMR gene database (ResFinder version 8 (1)) was used to annotate properly paired reads (MGmapper option fullmode) where each read pair had sequence coverage of at least 80% compared with the length of the trimmed reads. This was the only filter that was applied to discard a read pair. An AMR gene database (ResFinder) was used to annotate properly paired reads. 12 To enable multiple database hits, AMR genes were mapped using the fullmode option in MGmapper for the most optimal abundance calculation. Genomic annotation was performed by identifying the best hit (MGmaper option bestmode) for a pair of reads when aligned against a range of reference sequence databases. The total bacterial read count for a sample was calculated as the sum of read counts from each of the bacteria-related databases. The total fraction of unmapped reads for all samples was used to translate the percentage of unmapped reads into Z-scores (i.e., the number of standard deviations from the mean). Community profiling was performed using Kraken. 13 Relative abundance of AMR genes and bacterial genera was calculated as fragments per kilobase of transcript per million mapped reads (FPKM). This was done to account for both sample-wise sequencing depth differences and size-dependent probability of observing a reference. For AMR genes, FPKM was calculated on each individual ResFinder reference sequence. FPKMs were subsequently summed up across categories to bacteria genera, drug class level and highly homologous AMR gene groups. Alpha and beta diversity and richness were calculated on rarefied versions of the resistance and bacterial count matrices.
Results
Clinical data
Ten patients were successfully enrolled and randomised into two clinical trials (Table 1). Age ranged from 20 to 78 years old, and 6 of 10 patients were female. The bloodstream isolates encountered were Escherichia coli, Streptococcus agalactiae, Klebsiella pneumoniae and Citrobacter koseri. The trial antibiotics administered for directed therapy for bloodstream infection were ampicillin, ceftriaxone, cefazolin, benzylpenicillin, piperacillin-tazobactam and meropenem. Empiric antimicrobial therapy was heterogeneous among patients, and there was no washout period. These antibiotic regimens included ampicillin plus gentamicin, clindamycin plus benzylpenicillin, piperacillin-tazobactam, piperacillin plus gentamicin and ampicillin plus gentamicin plus metronidazole (Figure 1). One patient was pregnant (Patient 10). No patient received nutrition via an enteral feeding tube. The patient timeline of gut microbiome antimicrobial exposure and faecal sampling is outlined in Figure 1.
Summary of clinical data.


Patient timeline of gut microbiome antimicrobial exposure and faecal sampling.
Faecal sampling quality
Four patients produced four faecal specimens, three produced three specimens, two produced two specimens and one produced one specimen during their inpatient stay in the hospital whilst enrolled in the clinical trial. Of a total of 30 specimens, 15 were rectal swabs and 15 were stool samples. One patient had their stool specimens described as watery, whilst the rest were formed. Faecal sampling quality was determined for all 30 specimens (Table 2). Overall, DNA concentration was lower for rectal swabs compared to stool samples (16.35 vs 29.70 ng/µL). We determined that stool specimens had higher biomass than rectal swabs and, on average, yielded a much lower percentage of human (0.22% vs 24.78%) and skin flora (4.36% vs 16.37%) reads.
Quality of faecal specimens among patients enrolled in a clinical trial.
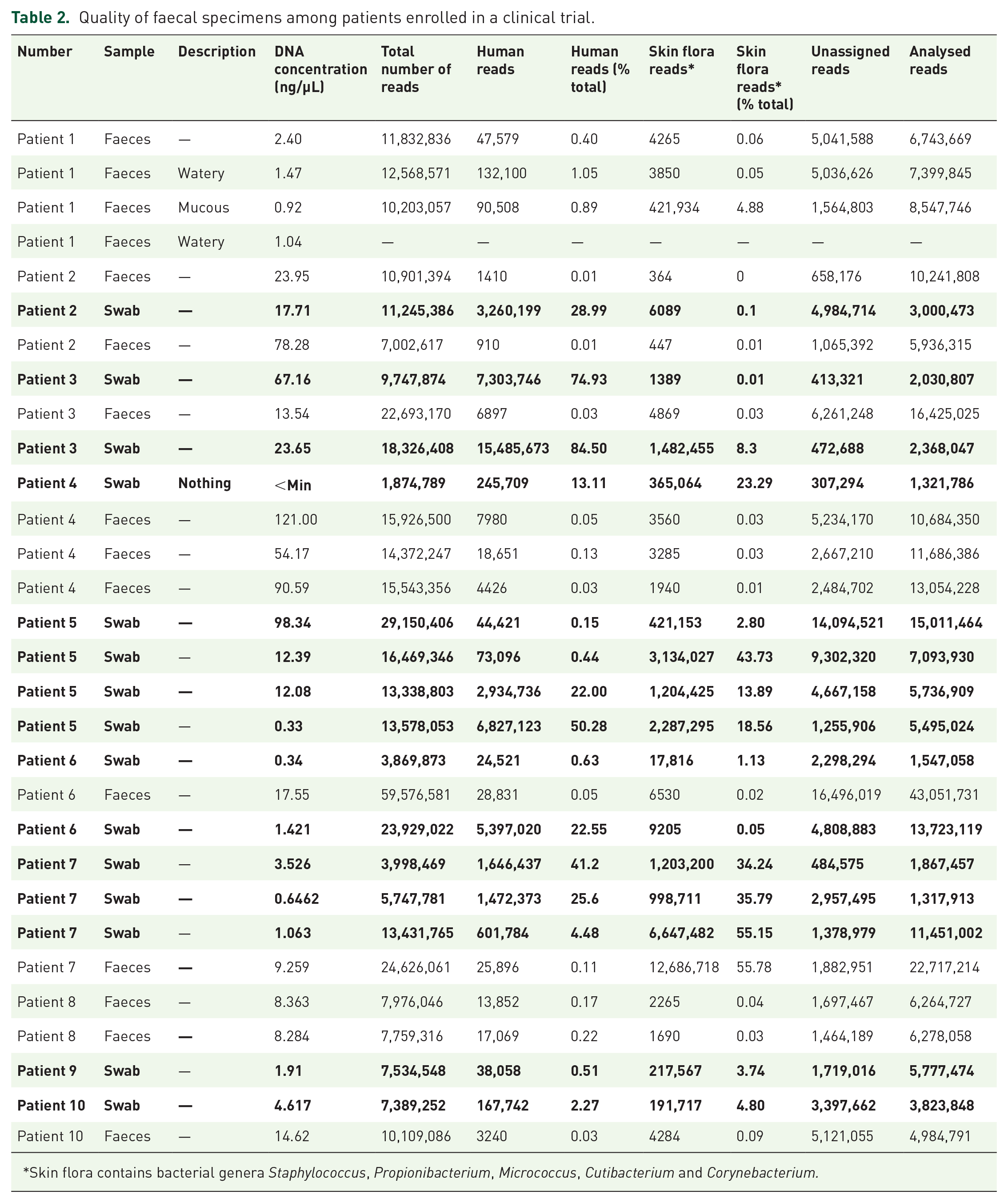
Skin flora contains bacterial genera Staphylococcus, Propionibacterium, Micrococcus, Cutibacterium and Corynebacterium.
Community profiling and antimicrobial resistance gene abundance
All stool specimens collected underwent community profiling and ARG abundance determination (Figures 2 and 3). Of trial participants receiving ampicillin (Patients 6, 8, 9 and 10), Patients 6, 9 and 10 demonstrated preservation of key anaerobic bacteria (Parabacteroides spp., Bifidobacterium spp., Bacteroides spp.). These patients also showed a marked reduction in Prevotella spp., Streptococcus spp., Lactobacillus spp. and Actinomyces spp. Enterococcus faecium became present or more abundant in Patients 8 and 10. Clostridioides difficile became more abundant in Patient 10. A reduction in the abundance of yeast (Saccharomyces spp.) was observed in Patient 8. An increase in abundance of blaTEM—1A was seen in Patient 6. Of trial participants receiving ceftriaxone (Patients 1 and 3), Patient 3 demonstrated a marked reduction in Enterobacterales (including E. coli) in addition to the beta-lactamase gene blaTEM-1A. Clostridioides difficile and E. faecalis became present or more abundant during antimicrobial administration. Patient 1 had no beta-lactamase genes detected and exhibited a marked reduction in anaerobic species (Bifidobacterium spp., Clostridium spp., Bacteroides spp.) in the last specimen collection which coincided with receipt of amoxicillin-clavulanate 24 h prior. There were no ESBL genes detected in trial participants receiving ceftriaxone. Of trial participants receiving piperacillin-tazobactam (Patient 7), E. faecium and yeast species (Candida spp. and Saccharomyces spp.) became present or increased in abundance, coincident with a reduction in key anaerobic species (Clostridium spp. and Bifidobacterium spp.) and the abundance of blaZ gene (Penicillinase of Staphylococcus aureus). Of trial participants receiving meropenem (Patient 5), there was an increased abundance of E. faecium and Bacteroides spp. with a coincident reduction in Enterobacterales and other anaerobic species (Parabacteroides spp., Clostridium spp. and Bifidobacterium spp.). Of note, this patient also received piperacillin-tazobactam for 3 days prior to commencement of meropenem. A marked reduction in the abundance of certain beta-lactamase genes (blaSHV-100, blaOXA-392, blaCMY-18) when transitioning from piperacillin-tazobactam to meropenem. A marked increase in abundance of the cfxA gene (encoding CfxA beta-lactamase) was also noted. There were no carbapenemase genes detected. Of trial participants receiving cefazolin (Patient 2), an increase in key anaerobic species (Bifidobacterium spp., Clostridium spp., Bacteroides spp. and Anaerococcus spp.) and abundance of blaTEM-1A was observed. Of trial participants receiving benzylpenicillin (Patient 4), an increased abundance of Enterobacterales and anaerobic species (Parabacteroides spp., Clostridium spp., Bacteroides spp.) and of certain beta-lactamase genes (blaZ, blaSHV) and the penA gene (encoding PLP2, which is associated with decreased susceptibilities to extended-spectrum cephalosporins in gonococcal strains). A reduction in Bifidobacterium spp., Lactococcus spp. and Streptococcus spp. was also seen in this patient. Of patients who received at least one dose empiric gentamicin (Patients 1, 2, 3, 6, 7, 8, 9 and 10), an increase in abundance of aminoglycoside resistance genes (aph(6)-Id, aph(3’)-Ia, aph(3’)-Ib, aac(3)-XI) were observed in faecal specimens following exposure.
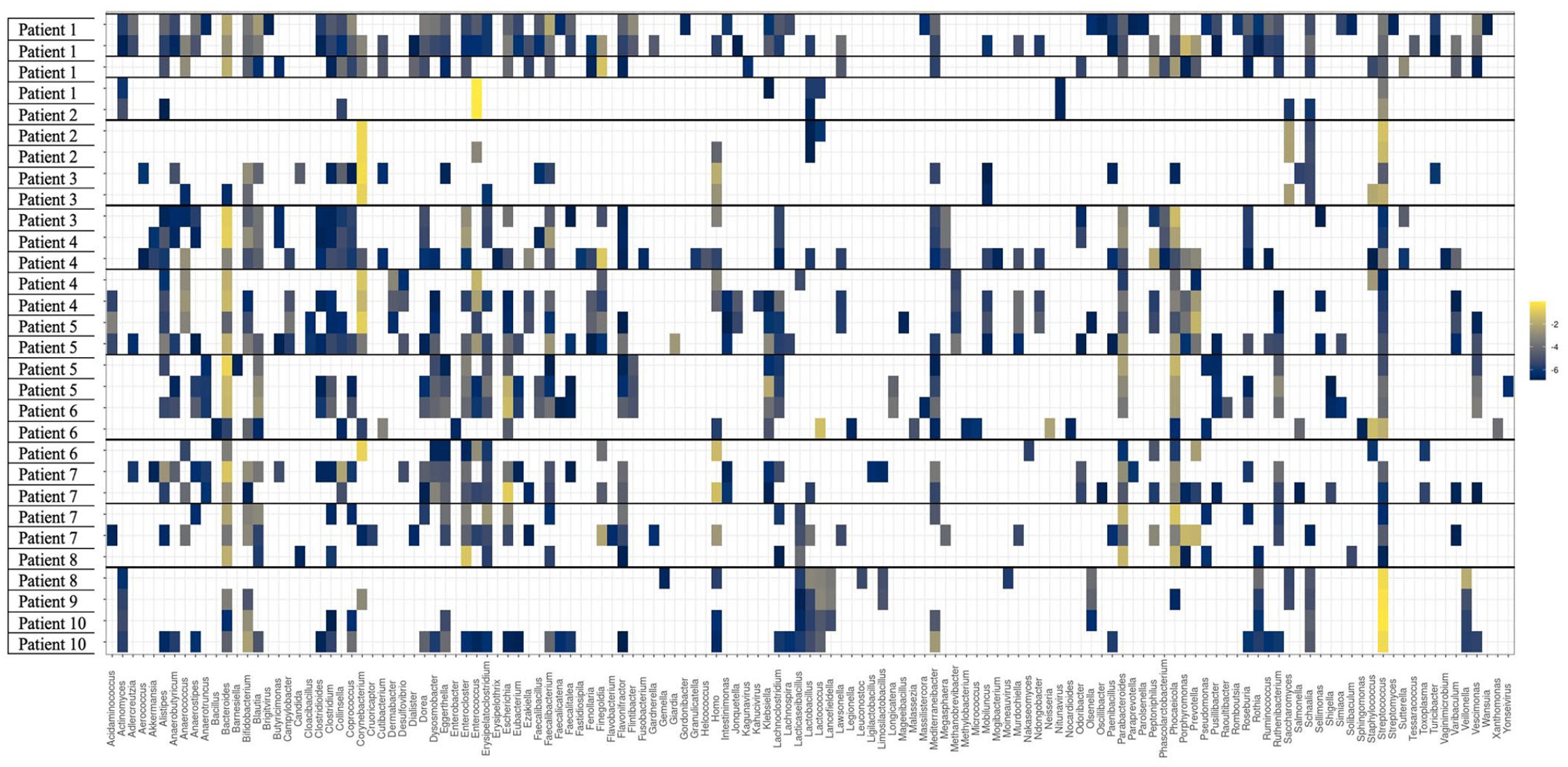
Community profiling of stool specimens collected from hospitalised patients enrolled in a clinical trial.
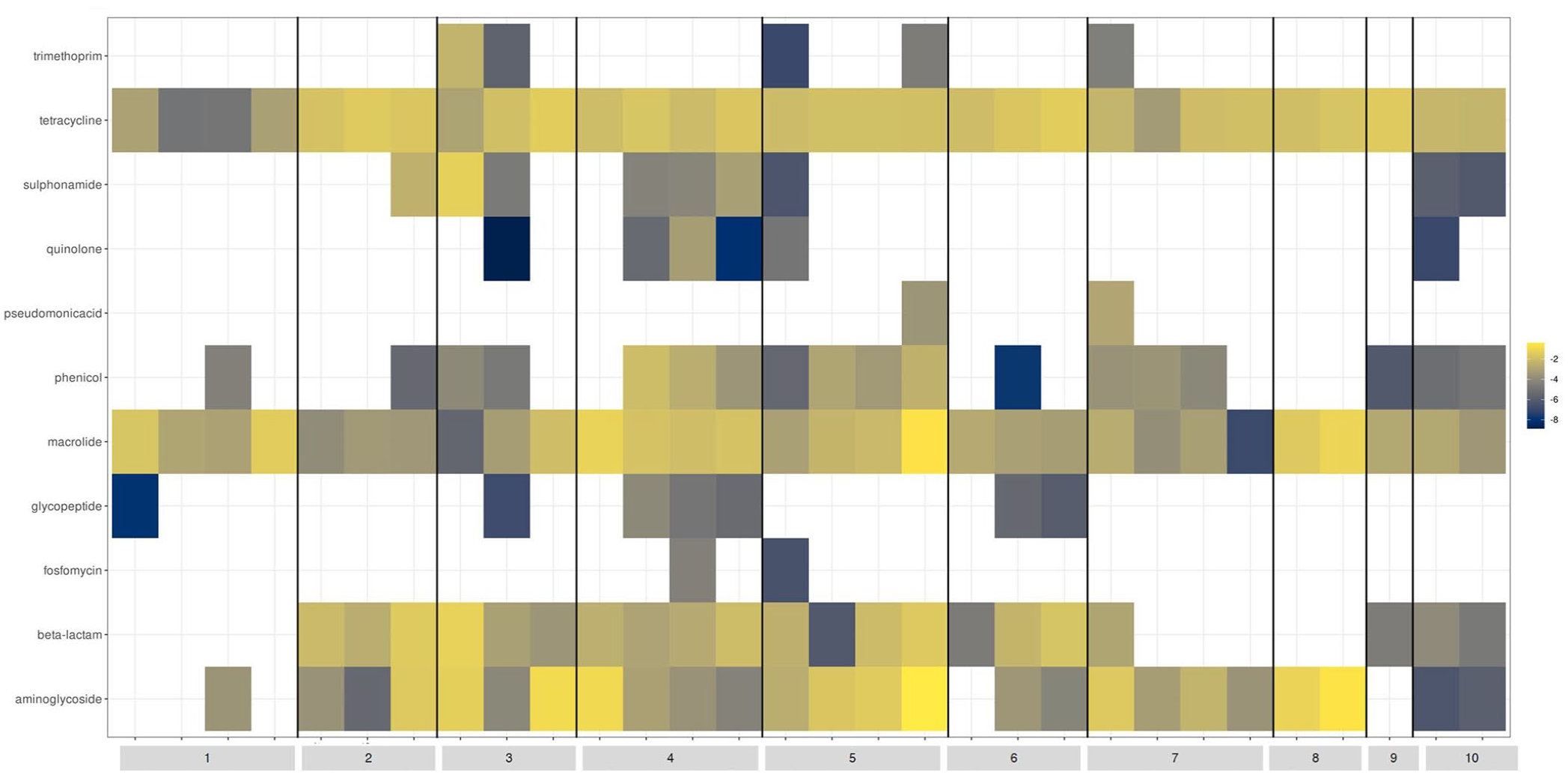
Antimicrobial resistance gene abundance in stool specimens collected from hospitalised patients enrolled in a clinical trial.
Diversity and richness
The Shannon index for alpha diversity was calculated for each specimen (Figure 4). The most dramatic decline in the Shannon index (4.286 to 1.880) was seen in Patient 5 receiving meropenem. Patients exposed to antimicrobials with negligible anaerobic activity (cefazolin, ceftriaxone; Patients 1, 2 and 3) demonstrated good preservation of diversity and richness indices. Similar high Shannon indices were noted for patients exposed to ampicillin, except for Patient 8 who received 2 days of piperacillin-tazobactam prior to enrolment into the clinical trial. Despite being exposed to metronidazole, Patient 10 had the highest recorded Shannon index values (both above 4.30).

Intra- and inter-patient differences in the Shannon index among clinical trial patients receiving different antimicrobials.
Discussion
This nested sub-study was undertaken from within two clinical trials and sought to identify key changes in vivo that occur in the gut microbiome on exposure to different antimicrobials in a controlled setting. In addition, we aimed to establish an appropriate workflow and methodology for integrating microbiome analysis into clinical trials. There was a large amount of variability in DNA concentration values between specimens, which likely reflected differences in specimen volume, collection technique, staff training, as well as specimen type. Optimisation of the pre-analytical phase of microbiome analysis would require stool samples collected by (or with direct oversight from) a trained trial coordinator. Because specimens were batch tested, we were unable to accurately assess the concentration of DNA yielded in relation to time from collection to DNA extraction. Enhanced degradation of nucleic acid resulting from improper or prolonged storage has been well documented in the literature.14,15
Our study was able to successfully link clinically important gut microbiome changes to individual antibiotic exposure in a clinical trial setting. Relative abundance of E. faecium, which is an ESKAPE pathogen (acronym of: E. faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa and Enterobacter species) responsible for many healthcare-associated infections 16 was observed to increase in individual patients that received ampicillin, meropenem and piperacillin-tazobactam. 16 There were no van genes detected. Although thought to cause comparatively less collateral damage, ampicillin may target specific anaerobes, Enterobacterales and E. faecalis, culminating in an environment that enables the expansion of E. faecium. 17 This finding has important antimicrobial stewardship and infection control implications and thus requires further investigation. C. difficile was detected in two patients, one who received a combination of ampicillin, gentamicin and metronidazole and the other with ceftriaxone. Although metronidazole has been described as a protective factor against C. difficile infection, colonisation resistance from ampicillin and gentamicin, favouring expansion of C. difficile, may have overcome this effect. 18 Pre-clinical models have been able to shed light on the precise gut microbiome perturbations that lead to C. difficile infection. 19 Porphyromonadaceae, Lachnospiraceae, Lactobacillus and Alistipes inhibit intestinal colonisation by C. difficile, whereas the genera Escherichia and Streptococcus have the opposite effect. 19 C. difficile was not detected from stool samples in patients receiving piperacillin-tazobactam or meropenem, although the analytical sensitivity of targeted molecular assays (e.g., singleplex real-time PCR) is likely to be greater than for metagenomic sequencing. 20 The differences between C. difficile nucleic acid detection (and quantitation) from metagenomic sequencing versus conventional testing (e.g., stool EIA and toxin PCR) with respect to clinical disease and infection control implications require investigation.
The differential depletion of anaerobic genera from the gut microbiome on exposure to antimicrobials with anti-anaerobic activity was notable. Bifidobacterium species are important to gut microbial diversity and were relatively preserved in patients exposed to ampicillin. Despite predictably depleting Clostridium and Bifidobacterium species, exposure to meropenem resulted in an increased abundance of Bacteroides species. Ambler class A beta-lactamase gene cfxA is associated with cephamycin resistance and is found on Bacteroides and Parabacteroides species. 21 The cfiA gene in B. fragilis is a metallo-beta-lactamase that is responsible for carbapenem resistance and has been documented. 22 There were no cfiA genes identified in our study.
With regards to beta-lactam antimicrobial resistance determinants, blaTEM-1A abundance increased in the setting of ampicillin exposure. Ampicillin likely selected TEM-1A-producing Enterobacterales in the gut microbiota. 23 This has direct and immediate clinical consequences in that further bloodstream or healthcare-associated infection would likely be due to a TEM-1A producer and would require therapy with a further detrimental effect on the gut microbiome (e.g., amoxicillin-clavulanate, ceftriaxone). This was made evident with Patient 3 who received ceftriaxone and demonstrated a rapid reduction in abundance of blaTEM-1A followed by a rise in C. difficile and E. faecalis. Patient 5 received piperacillin-tazobactam in the days prior to clinical trial enrolment, followed by meropenem. The first faecal specimen at the point of antimicrobial transition demonstrated an abundance of beta-lactamase genes (blaSHV-100, blaOXA-392 and blaCMY-18), which were eradicated with subsequent meropenem exposure. Concerns over the clinical efficacy and in vitro activity of piperacillin-tazobactam against OXA-1 and CMY-2-producing Enterobacterales have been raised.24,25 Also, exposure of the gut microbiome to a certain class of antibiotics may result in resistance development to a separate class of antibiotics (i.e., cross-resistance). This has been demonstrated in a pre-clinical model. 26
Yeast species (Candida spp. and Saccharomyces spp.) became most prevalent in the gut microbiome in the patient with piperacillin-tazobactam exposure. Although many broad-spectrum antibiotics have been associated with gastrointestinal tract colonisation with Candida species, piperacillin-tazobactam may be associated with a much higher comparative risk. 27 Monitoring the risk of invasive candidiasis with piperacillin-tazobactam exposure in a clinical trial setting is important.
Integration of gut microbiome analysis into contemporary antibiotic clinical trials has been proposed to better aid treatment decisions and guide the placement of new antimicrobials in the clinical space in the post-marketing phase of development. 8 A microbiome nested sub-study was performed in the PIRATE trial, which assessed the non-inferiority of shorter antibiotic courses (7 vs 14 days) for Gram-negative bacteraemia. 28 Enrolled participants were followed up to 30 days, and there was no difference in ARG abundance (median counts per million) or microbial species diversity (Shannon species diversity) between the two groups. In healthy volunteers, gut microbiome diversity and richness have been reported in a study to return to pre-antibiotic treatment levels after approximately 2 months. 4 Long-acting antibiotics (e.g., azithromycin) and pre-treatment microbiota resembling sick and hospitalised patients lead to more persistent antibiotic-related microbiome changes. Other studies have echoed the importance of pre-treatment microbiota composition. 29 Moreover, 20% of patients in our study were taking a proton pump inhibitor, which has been independently associated with lower abundance in gut commensals and lower microbial diversity. 30 This likely confounded findings in these patients.
There were many limitations to this study. First, the evaluable population was heterogeneous and was derived from two separate clinical trials that sought to answer two different clinical questions. Moreover, there were many trial antibiotics administered (six in total), with many having pre-trial exposure to different antimicrobials altogether. None of the patients received only a single antimicrobial, and the flow-on effect from a previous antimicrobial agent could have contributed to findings. In addition, no patients had a faecal specimen taken prior to antibiotic treatment, thus limiting our interpretations of the data. Although this represents a “real-world” setting, it creates difficulty in defining true antibiotic-associated effects on the gut microbiome. Second, faecal sampling number and duration were suboptimal, and specimens were batch tested. Gut microbiome adaptations to antimicrobials are known to evolve over time, which requires analysis over weeks to months to get an accurate picture. From a feasibility standpoint, it is much easier to collect faecal specimens from patients admitted to the hospital. This needs to be weighed up with every study where collecting quality stool samples from the community may cause additional issues with the study (i.e., participant response rates, storage and transport of specimens and additional costs). A proposed solution to this has been the development of commercial self-collection kits with in-built DNA/RNA stabilisers, which prevent nucleic acid degradation and enable prolonged storage and transport times. Third, half of all specimens collected were from rectal swabs, which proved to produce more human and skin flora sequences using the techniques and swab type in this study. Although easier to collect and perhaps more acceptable to patients, this needs to be weighed against study quality. Fourth, this study only recruited 10 patients and therefore did not have enough power to detect gut microbiome effects that may be evident with certain antimicrobials. By having large groups of individuals being exposed to the same antimicrobial in a clinical trial setting, this would allow for statistical analysis, which could result in a more refined understanding.
Conclusion
Understanding the determinants of antimicrobial resistance development at an individual level is of great clinical importance. Stool samples provide higher biomass samples for sequencing and should be considered the preferred specimen type obtained for these studies. A number of challenges exist with regards to integrating gut microbiome analysis into clinical trials (e.g., obtaining an antibiotic-free stool specimen at baseline). Narrow-spectrum antibiotics may have a greater, and more clinically important, effect on our gut microbiota than once thought. The presence of key anaerobes such as Clostridium and Bifidobacterium species may play an important role in colonisation resistance and may be suitable outcome measures for future clinical trials. Further refinement of clinical trial infrastructure, methodology and workflow is required before existing challenges can be overcome.