
Abstract
Fostemsavir, a prodrug of the first-in-class gp120-directed attachment inhibitor temsavir, is indicated in combination with other antiretrovirals for the treatment of multidrug-resistant HIV-1 in adults who are heavily treatment-experienced (HTE). Temsavir binds to HIV-1 gp120, close to the CD4 binding site, preventing the initial interaction of HIV-1 with CD4 on the host cell. Amino acid substitutions at four positions in gp120 have been identified as important determinants of viral susceptibility to temsavir (S375H/I/M/N/T/Y, M426L/P, M434I/K, M475I), with a fifth position (T202E) recently described. For most currently circulating group M HIV-1 subtypes, the prevalence of these resistance-associated polymorphisms (RAPs) is low. As with many other antiretrovirals, the impact of RAPs is modified by other changes in the target molecule. Different regions of gp120 interact to modify the temsavir binding pocket, with multiple amino acids playing a role in determining susceptibility. Extensive variability of HIV-1 gp120 means the susceptibility of clinical isolates to temsavir is also highly variable. Importantly, in vitro measurement of the susceptibility of clinical isolates to temsavir does not necessarily capture the range of susceptibilities of the heterogeneous mix of viruses generally present in each isolate. Due to these factors and limited phenotypic clinical data, thus far, no relevant phenotypic cutoff or genotypic algorithms have been derived that reliably predict response to fostemsavir-based therapy in individuals who are HTE; therefore, pre-treatment temsavir resistance testing may be of limited benefit. In the phase III BRIGHTE study, re-suppression after virologic failure was observed in some participants despite treatment-emergent genotypic and/or phenotypic evidence of reduced temsavir susceptibility, and substantial CD4+ T-cell count increases occurred even among participants with HIV-1 RNA ⩾40 copies/mL at Week 240. Clinical management of people who are HTE and experience virologic failure during treatment with fostemsavir-based regimens requires an individualized approach with consideration of potential benefits beyond virologic suppression.
Introduction
Fostemsavir (Rukobia; ViiV Healthcare, Durham, NC), a prodrug of the first-in-class gp120-directed attachment inhibitor temsavir, is indicated for use in combination with other antiretrovirals (ARVs) for the treatment of multidrug-resistant HIV-1 in adults who are heavily treatment-experienced (HTE).1–7 Temsavir binds to HIV-1 gp120, close to the CD4 binding site, locking the molecule into a “closed” conformational state that prevents the initial interaction of HIV-1 with the host CD4+ T cell and subsequent HIV-1 binding and entry.8–11 With this unique mechanism of action, temsavir is active against both CCR5- and CXCR4-tropic viruses and shows no cross-resistance with other ARV classes, including other viral entry inhibitors, with the possible exception of context-dependent cross-resistance with ibalizumab.12–16 The clinical efficacy and safety of fostemsavir have been demonstrated in a phase IIb study, in combination with raltegravir and tenofovir disoproxil fumarate (TDF), in adults who were treatment-experienced (AI438011),4,6 and in a phase III study, in combination with optimized background therapy (OBT), in adults who were HTE with limited treatment options (BRIGHTE).1–3,5,7 Results through Week 240 of the BRIGHTE study showed that fostemsavir 600 mg twice daily plus OBT resulted in durable virologic suppression and clinically meaningful improvements in CD4+ T-cell count and CD4+/CD8+ ratio in a population with advanced disease and multidrug-resistant HIV-1. 2
People with HIV-1 who are HTE make up a heterogeneous population who often face multiple clinical and social challenges, including multidrug-resistant virus, cumulative drug toxicity, comorbidities, polypharmacy, and barriers to treatment adherence and retention in care.17–23 With no consensus on the definition of HTE status, reports of prevalence range from 2% to 16%.19–22 Treatment regimens for individuals who are HTE should optimally include well-tolerated ARVs with mechanisms of action distinct from previously existing classes and few drug–drug interactions. 23 The assessment of new ARV agents and/or strategies for this population is complicated by the range of individualized treatment needs and the lack of a standardized regimen to act as a control. 17 This presents challenges in designing experiments with a placebo group and has resulted in variable and complex study designs that can be difficult to interpret. 17 Nevertheless, as of January 2025, there are three first-in-class ARVs indicated for the treatment of multidrug-resistant HIV-1 in individuals who are HTE: fostemsavir (first approved in the United States in July 2020), 24 ibalizumab (a CD4-directed post-attachment inhibitor, first approved in the United States in March 2018), 25 and lenacapavir (an HIV-1 capsid inhibitor, first approved in the European Union in August 2022 26 ). 23 Constructing treatment regimens that incorporate these new classes of ARV requires an individualized approach with careful consideration of multiple factors to achieve maximum benefit. In a population in which there is already a high frequency of multidrug-resistant HIV-1, minimizing the risks and consequences of further drug resistance is an important consideration. 23
Specific amino acids at four HIV-1 gp120 positions surrounding the temsavir binding site (375, 426, 434, and 475) are the most important determinants of viral susceptibility to temsavir.1,7,9,13,14,27,28 Substitutions at gp120 amino acid positions 116(P), 202(E), and 204(D) have also been reported to have a measurable impact on susceptibility to temsavir, but these are extremely rare in the population.14,16,29,30 For most currently circulating HIV-1 subtypes, the prevalence of resistance-associated amino acid polymorphisms (RAPs) at positions 375, 426, 434, and 475 is low (<10%), and most (~80%) clinical isolates from individuals naive to fostemsavir show a high degree of susceptibility to temsavir, with half-maximal inhibitory concentrations (IC50s) of <10 nM.7,29,31 The exceptions to this are group O viruses (consensus H375), group N viruses (consensus M375, L426, I434), and the circulating recombinant form CRF01_AE (consensus H375, I475), which show high levels of reduced susceptibility to temsavir.14,29 The extensive variability of HIV-1 gp120 means that in vitro measurements of temsavir susceptibility vary widely among clinical isolates of HIV-1 both with and without RAPs or resistance-associated substitutions (RASs).14,27,31,32 In this narrative review, we describe the mechanisms and context dependency of reduced susceptibility to temsavir and summarize available data on the emergence and potential consequences of genotypic and phenotypic changes in gp120 during treatment with fostemsavir in clinical trials.
Mechanisms of reduced susceptibility to temsavir
Temsavir mechanism of action
HIV-1 gp120, the target of temsavir, is a complex, heavily glycosylated protein crucial for the process of viral attachment and entry into host cells.33–35 Situated on the viral surface, with highly conserved host protein binding sites that are exposed during the viral entry process, gp120 is a primary target for humoral immune responses. To avoid humoral responses, HIV has developed extensive variability in overall gp120 amino acid sequence and glycosylation patterns and a high degree of structural heterogeneity and conformational flexibility that masks conserved receptor-binding sites.33–36 Because of this, gp120 can be a difficult molecule to target with ARVs. 35
Modeling and crystallographic studies suggest that temsavir binds to gp120 in a structurally conserved hydrophobic pocket below and on the opposite side of the β20-21 loop to that recognized by CD4 (Figure 1).8,9 This binding acts to inhibit HIV-1 entry into host cells through two mechanisms of action: at higher concentrations, temsavir binding allosterically interferes with CD4 binding; at lower concentrations, temsavir binding stabilizes the molecule in a “closed” or “State 1” conformation that prevents the CD4-induced conformational rearrangements required for the next steps in the viral entry process.8–10,33,37 These mechanisms prevent the initial interaction of gp120 with the host cell, thus inhibiting subsequent HIV-1 binding and entry.8–10,37

Chemical structures of (a) fostemsavir and (b) temsavir and homology models of (c) CD4 (protein data bank code 2NXY), in brown, and (d) temsavir (protein data bank code 5U7O), in magenta, bound to wild-type HIV-1 JR-FL gp120, in green, with annotation to show key amino acid positions. Models illustrate the distinct CD4 and temsavir binding sites on opposite sides of the β20-21 loop (in blue).8,9,38
Amino acid substitutions associated with reduced susceptibility to temsavir
Extensive preclinical studies with temsavir and related experimental attachment inhibitors (BMS-378806 and BMS-488043), and genotypic and phenotypic analysis of clinical isolates of HIV-1 from multiple sources, including clinical trials of fostemsavir, initially identified four amino acid positions in conserved regions of HIV-1 gp120 that play an important role in reducing the susceptibility of the virus to temsavir: positions 375, 426, 434, and 475 (Table 1 and Figure 1).1,14,27,28,32,39,40 Amino acid substitutions L116P and A204D were also shown to decrease temsavir susceptibility in vitro; however, changes at these positions have not been observed in vivo in fostemsavir clinical studies, and both 116P and 204D are very rare among circulating subtypes of HIV-1 (only four and five of 10,733 full-length sequences, respectively, in the Los Alamos National Laboratory (LANL) HIV sequence database).14,29,30 Based on these studies, the amino acid substitutions (relative to HIV-1 HXB2) S375H/I/M/N/T, M426L/P, M434I/K, and M475I were pre-specified for monitoring in the BRIGHTE study.1,7,27 In an HIV-1 LAI background, most of these identified RAPs individually result in less than 100-fold reductions in susceptibility to temsavir, with double amino acid substitutions being required to achieve greater than 1000-fold reductions (Tables 1 and 2). Subsequently, S375Y was identified as a novel RAP present at baseline in two participants in BRIGHTE (although there are only 12 examples in the LANL database) and found to be associated with a substantial decrease in temsavir susceptibility when introduced into an HIV-1 LAI background.1,2,27,30 After further analysis of data from BRIGHTE, the amino acid substitutions S375H/I/M/N/Y, M426L, and M434K were designated as “most relevant” based on their impact on temsavir susceptibility and/or association with an HIV-1 RNA reduction of <0.5 log10 copies/mL from Day 1 to Day 8 of functional fostemsavir monotherapy (Table 1). More recently, gp120 amino acid position 202 has also been reported to have an impact on temsavir susceptibility. Although previous studies supported a lack of cross-resistance between temsavir and the CD4-directed post-attachment inhibitor ibalizumab, 13 two envelope clones from the Monogram Biosciences collection have been identified that show reduced susceptibility to both agents. 16 In one of these clones, 202E was shown to be a key substitution resulting in reduced susceptibility to temsavir and ibalizumab. 16 Like 116P and 204D, 202E is rarely observed in clinical isolates of HIV-1, with only two examples in the 2022 version of the LANL database. 30
Impact of gp120 amino acid substitutions introduced by site-directed mutagenesis on in vitro susceptibility to temsavir of HIV-1 LAI (subtype B) in a cell–cell fusion assay and HIV-1 JR-FL in a pseudovirus assay.

Amino acid substitutions numbered according to the HIV-1 HXB2 reference sequence. In the phase III BRIGHTE study, substitutions shown in bold font were pre-specified for monitoring at all time points (S375Y was pre-specified for monitoring from Week 240), and substitutions marked with * were considered to be most relevant based on impact on temsavir susceptibility or association with an HIV-1 RNA reduction of <0.5 log10 copies/mL from Day 1 to Day 8 of functional fostemsavir monotherapy in clinical studies.
Fold change in temsavir IC50 between wild-type HIV-1 LAI reference virus and HIV-1 LAI with the single amino acid substitution indicated in the first column in a cell–cell fusion assay.1,7,14,28
Fold change in temsavir IC50 between wild-type HIV-1 JR-FL reference virus and HIV-1 JR-FL with the single amino acid substitution indicated in the first column in a pseudovirus assay. 41
Frequency among 10,733 HIV-1 envelope sequences in the LANL HIV sequence database through December 31, 2022. 30
In a cloned envelope from the Monogram Biosciences library, E202 was associated with a >400-fold reduction in temsavir susceptibility compared with T202 in the same background. 16
PDVF was defined as the following: before Week 24, confirmed (or last available before discontinuation) plasma HIV-1 RNA ⩾400 copies/mL after prior confirmed suppression to <400 copies/mL or >1 log10 copies/mL increase in HIV-1 RNA at any time above nadir, where nadir is ⩾40 copies/mL; at or after Week 24, confirmed (or last available before discontinuation) HIV-1 RNA ⩾400 copies/mL. 2
CRF01_AE sequences contributed most of the instances of 375H (72%; 1081/1511) and 475I (73%; 841/1152) in the LANL database.
29/66 fostemsavir-treated participants who met criteria for resistance testing had a temsavir susceptibility result, 13/29 had virus with a >3-fold increase in FC-IC50 from baseline, and population gp120 sequencing was successful for 11/13 participants. 28
Virologic response was defined as a maximal decline in plasma HIV-1 RNA of ⩾1 log10 copies/mL from Day 1 to Day 8. 40
FC, fold change; IC50, half-maximal inhibitory concentration; LANL, Los Alamos National Laboratory; NR, not reported; PDVF, protocol-defined virologic failure; SDM, site-directed mutagenesis; TDF, tenofovir disoproxil fumarate.
Impact of gp120 amino acid substitutions at positions 375, 426, 434, and 475 introduced by site-directed mutagenesis into two different viral envelope clones.

Amino acid substitutions numbered according to the HIV-1 HXB2 reference sequence.
Fold change in temsavir IC50 in a cell–cell fusion assay between wild-type reference virus and the same virus with the indicated amino acid substitution(s).
FC, fold change; IC50, half-maximal inhibitory concentration.
The effects of amino acid substitutions at positions 375, 426, 434, and 475 on temsavir susceptibility are consistent with the proposed binding site and mechanism of action.8–10 All four positions reside in or close to the modeled temsavir binding site. 8 Leucine and isoleucine have branched side chains that are predicted to sterically reduce the size of the temsavir binding site at positions 426 and 475, respectively.8,9 Although direct interactions between temsavir and S375 are minimal, bulky amino acid side chains at this position extend into the temsavir binding site and physically interfere with temsavir binding. 10 Prévost et al. 10 showed that in a subtype B HIV-1 envelope (JR-FL) the size of amino acid residue at position 375 correlated with the activity of temsavir in a single round neutralization assay; for the smallest amino acids (serine (S) or threonine (T)), temsavir IC50s were <1 nM, while for the largest amino acids (phenylalanine (F), tyrosine (Y), histidine (H), or tryptophan (W)), IC50s were >100 nM. M434 packs against the azaindole ring of temsavir bound to gp120, and the branched side chain of isoleucine at this position would require an alteration of the orientation of temsavir in the binding pocket. 9 In a biophysical analysis, the affinity (equilibrium dissociation constant (KD)) of temsavir for JR-FL gp120 molecules carrying RAPs (either alone: S375H/I/M/N, M426L, M434I, M475I, or in combination: S375H + M475I) varied from 0.7- to 74-fold compared with wild-type JR-FL gp120. 41 There was a strong correlation between temsavir IC50 in a pseudovirus assay and temsavir affinity and binding on-rate (r = −0.8940; p = 0.0011; Table 3). Importantly, for all tested gp120 molecules, temsavir was still able to completely block CD4 binding in a competition assay (at concentrations of 20 × KD for M434I, S375H, S375M, S375N, and S375H + M475I or 40 × KD for M426L, M475I, and S375I). 41 Amino acid position 202 is also located next to the temsavir binding site in gp120 and a glutamic acid (E) at this site likely sterically hinders temsavir binding. 16 Ionic interactions may also play a role since the virus carrying gp120 202K (lysine), a positively charged amino acid with a longer side chain than the negatively charged glutamic acid, retains susceptibility to temsavir. 16
Comparison of the on-rate (ka) of temsavir binding to gp120 carrying the indicated amino acid substitutions versus temsavir IC50 for pseudotyped viruses with the same amino acid substitutions. 41

IC50, half-maximal inhibitory concentration.
Given that temsavir binds to gp120 in a conserved region close to the CD4 binding site, it might be expected that amino acid substitutions that impact temsavir binding could also impact virus infectivity or “fitness.” Indeed, as previously noted, the substitutions that have the greatest individual impact on temsavir susceptibility in vitro (>100-fold, L116P, T202E, A204D, S375Y, and M434K) are all extremely rare (⩽0.1% in the LANL HIV sequence database) and have infrequently been observed in fostemsavir clinical studies.2,29 In BRIGHTE, there have been no reports of L116P or A204D, one case each of T202E and M434K, and two cases of S375Y.
Context dependency of temsavir susceptibility
While the key amino acids described above clearly result in measurable reductions in temsavir susceptibility, their impact in clinical isolates is highly variable. This suggests that the effects of changes at these positions are highly dependent on other polymorphisms in the HIV envelope (the envelope context), in which they occur, as has been described for neutralizing antibodies.16,31,42
When RAPs at positions 375, 426, 434, and 475 were introduced by site-directed mutagenesis into two different viral envelope clones (the subtype B laboratory strain LAI and an envelope derived from a subtype B clinical isolate from a participant in the phase IIa clinical study), several of the inserted amino acid substitutions had different effects on temsavir susceptibility in the two envelopes (Table 2; M. Gartland, personal communication). This was true for single substitutions (S375H/I/M/N) and double substitutions (S375M + M475I, M426L + M475I). Even within single clinical isolates (derived from participants in the phase IIa fostemsavir study), multiple clones, all carrying the same RASs in gp120, had in vitro temsavir susceptibilities spanning a 2- to 3-log range (Figure 2). 14 For example, 12 functional clones from participant 12, each containing S375H, had fold change (FC)-IC50s ranging from 7 to 1138, while the population assay gave a FC-IC50 of 19. Similar results were seen for participant 21, where all 30 clones contained M426L. Even for viruses with high (participant 27) or low (participant 16) levels of temsavir susceptibility, there was a wide range of FC-IC50s seen across multiple clones. 14

Variability of in vitro susceptibility to temsavir among individual clones from the same clinical isolate population with the same key amino acid substitutions. Clinical isolates were derived from participants at baseline in the phase IIa fostemsavir clinical study.
In an investigation into the correlates of temsavir resistance in subtype CRF01_AE (which carries naturally occurring, conserved H375 and I475), wide ranges of in vitro temsavir susceptibility within groups of clinical isolates of various subtypes based on the amino acid at position 375 were observed (Figure 3(a)). 10 Furthermore, introducing an H375S into CRF01_AE viruses did not completely restore temsavir susceptibility and, in addition to I475, amino acids at five other co-evolved positions within the inner gp120 domain layers (H61, Q105, V108, N474, and K476) were found to be involved in the observed lack of susceptibility to temsavir in CRF01_AE viruses. Further structure–function analyses showed that the temsavir binding pocket in gp120 is shaped by cooperation between distal regions of gp120 and that temsavir is able to adjust its conformation in the binding pocket to accommodate minor changes in Env conformation.
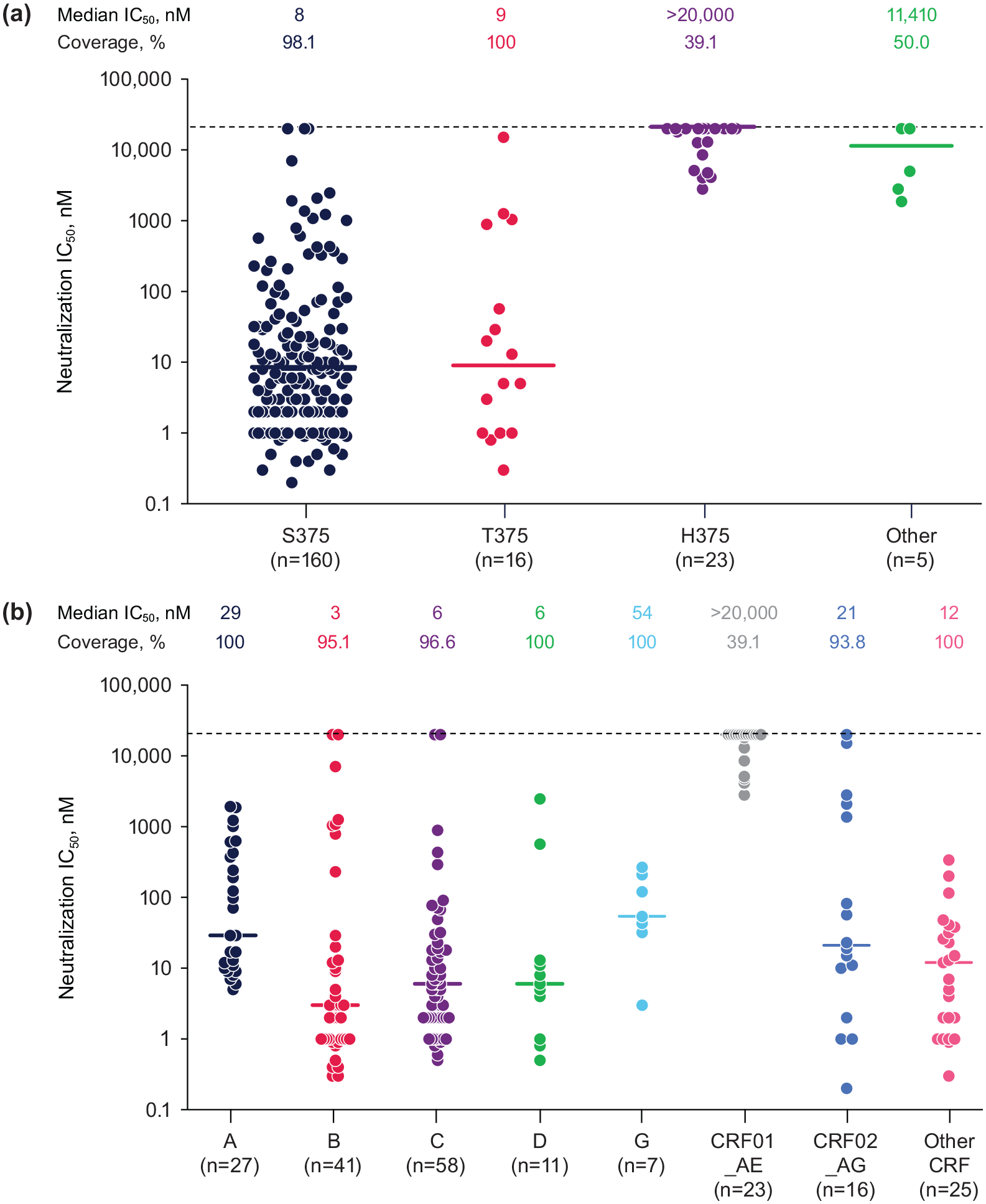
Variability of in vitro susceptibility to temsavir among different clinical isolates of HIV-1 (N = 208 biologically independent viral strains) grouped by (a) identity of the polymorphic residue at position 375 of gp120 and (b) virus subtype. Horizontal lines indicate median IC50, dotted line indicates maximum detectable IC50, % coverage indicates percentage of tested isolates with measurable IC50 within detectable range.
Another element to consider for context dependency of temsavir susceptibility is glycosylation. Potential N-linked glycosylation sites in gp120 have been shown to play a role in reduced susceptibility to the CD4-directed post-attachment inhibitor ibalizumab.16,43 In studies assessing the impact of gp120 202E on reduced susceptibility to temsavir and ibalizumab, loss of a highly conserved N-linked glycosylation site close to position 202, through the introduction of the substitution N197D, resulted in complete restoration of susceptibility to ibalizumab and partial restoration of susceptibility to temsavir. 16
Virologic failure in clinical studies of fostemsavir
Phase IIb study
In the phase IIb study (NCT01384734) in adults with HIV-1 who were treatment-experienced, fostemsavir at doses of 400 mg twice daily, 800 mg twice daily, 600 mg once daily, or 1200 mg once daily was well tolerated and resulted in similar antiviral efficacy and immunologic responses relative to the active comparator, ritonavir-boosted atazanavir (both in combination with raltegravir and TDF), through Weeks 24 and 48.4,6,28 Eligibility criteria for this study included a clinical isolate temsavir IC50 of <100 nM at baseline; nevertheless, amino acid polymorphisms at positions 375, 426, 434, or 475 were detected at baseline in 42% of participants in the fostemsavir group.4,28 There was no correlation between the presence of these baseline polymorphisms and meeting on-treatment resistance testing criteria through Week 48. 28 Among 66 participants in the fostemsavir group who met criteria for resistance testing, 29 had a successful temsavir susceptibility result (using the PhenoSense® Entry assay, Monogram Biosciences, South San Francisco, CA, USA), and for 13/29, there was a >3-fold increase in FC-IC50 from baseline. 28 Population sequencing of gp120 was successful for samples from 11 of these 13 participants, 7 of which showed emergent changes at positions 375, 426, or 434, most commonly M426L (n = 5). 28 Emergence of reduced susceptibility to temsavir did not preclude re-suppression; among 13 participants for whom decreased susceptibility to temsavir was detected during treatment, five achieved re-suppression to HIV-1 RNA <50 copies/mL during the study, regardless of S375S/N and M426L substitutions. 28
Phase III BRIGHTE study
In the ongoing phase III BRIGHTE study (NCT02362503), fostemsavir 600 mg twice daily plus OBT has been well tolerated and has demonstrated durable virologic and immunologic responses through 240 weeks of treatment in adults who were HTE with advanced disease and limited treatment options (⩽2 non-investigational ARVs available).2,3,5 BRIGHTE enrolled participants in two cohorts: a Randomized Cohort (RC) of participants with one or two fully active ARVs that could be included in the OBT, and a compassionate-use Non-randomized Cohort (NRC) of participants who had no remaining fully active approved ARVs at the beginning of the study. At baseline, most participants (85%; 316/371) had received at least five prior ARV regimens, 86% (320/371) had a history of AIDS, and 30% (112/371) had CD4+ T-cell count <20 cells/mm3. 44
The presence of RAPs at baseline may explain some of the differences in virologic response in the BRIGHTE study. In a subset of participants without evidence of any residual activity from their failing regimen, the baseline presence of at least one RAP designated as “most relevant” (S375H/I/M/N/Y, M426L, M434K) was significantly associated with lower odds of a Day 8 virologic response (>0.5 log10 copies/mL decrease in HIV-1 RNA). 45 At Week 192, 53% (145/272) of participants in the RC achieved Snapshot virologic response, and the presence of at least one “most relevant” RAP was associated with significantly lower odds of virologic response. 46
Rates of protocol-defined virologic failure (PDVF) through Weeks 96 and 240 were as expected in this population with multidrug-resistant HIV-1: 23% (63/272) and 29% (80/272), respectively, in the RC and 49% (49/99) and 54% (53/99), respectively, in the NRC.2,27 Protocol-defined virologic failure was most frequent among participants with baseline CD4+ T-cell count <20 cells/mm3 and a history of AIDS (Table 4). 2 Among the 133 participants with PDVF through Week 240, 122 had available population genotypic and phenotypic temsavir susceptibility results. 2 Pre-specified RASs were emergent in samples from 52% (n = 63) of participants, with S375N or M426L being the most frequent (Table 5). In most cases (89%; 56/63), emergent pre-specified RASs were associated with a >3-fold reduction in in vitro temsavir susceptibility relative to the baseline sample. There were also nine cases of a >3-fold reduction in temsavir susceptibility in the absence of detectable emergent pre-specified RASs. Thus, 59% (72/122) of participants with PDVF had detectable genotypic or phenotypic evidence of a change in temsavir susceptibility from baseline. Notably, like the phase IIb study, these emergent changes did not preclude virologic re-suppression. Among 75 participants with treatment-emergent RASs or >3-fold change in temsavir susceptibility who remained in the study after PDVF, 20% (n = 15) had HIV-1 RNA <40 copies/mL, seven of whom had no changes to their OBT regimen. Importantly, substantial increases in CD4+ T-cell count were observed even among study participants with HIV-1 RNA ⩾40 copies/mL at Week 240 (mean increase, 251 cells/mm3). 2
Incidence of PDVF by baseline characteristics through Week 240. 2

PDVF, protocol-defined virologic failure (before Week 24: confirmed (or last available before discontinuation) plasma HIV-1 RNA ⩾400 copies/mL after prior confirmed suppression to <400 copies/mL or >1 log10 copies/mL increase in HIV-1 RNA at any time above nadir, where nadir is ⩾40 copies/mL; at or after Week 24: confirmed (or last available before discontinuation) HIV-1 RNA ⩾400 copies/mL).

Sequencing results at additional on-treatment time points around the time of PDVF are included where available (not limited to only the PDVF time point).
Denominator for proportions with substitutions is the number sequenced for each cohort and overall.
S375H/I/M/N/T, M426L, M434I/K, M475I.
S375H/I/M/N/Y, M426L, M434K.
PDVF, protocol-defined virologic failure (before Week 24: confirmed (or last available before discontinuation) plasma HIV-1 RNA ⩾400 copies/mL after prior confirmed suppression to <400 copies/mL or >1 log10 copies/mL increase in HIV-1 RNA at any time above nadir, where nadir is ⩾40 copies/mL; at or after Week 24: confirmed (or last available before discontinuation) HIV-1 RNA ⩾400 copies/mL).2
Polymorphisms and temsavir susceptibility in global clinical isolates
Data from the BRIGHTE study, in which most participants had HIV-1 subtype B, show that high in vitro temsavir FC-IC50s and/or RAPs at baseline in some cases did not preclude a virologic response to treatment with fostemsavir-based regimens. 29 This may in part be due to the relatively high 600-mg twice-daily fostemsavir dose, which was selected based on exposure-response modeling that incorporated the variability in temsavir pharmacokinetics and baseline protein binding–adjusted temsavir IC50 observed in the phase IIa and IIb studies. 3 Nevertheless, it is important to be aware of natural variations in temsavir susceptibility and prevalence of RAPs that could affect the efficacy of fostemsavir in the global population with HIV-1. 29
Wide variability in susceptibility to temsavir has been observed overall and within subtypes (Figures 3(b) and 4). In an analysis of 1337 individual envelopes tested using the PhenoSense Entry assay, IC50s ranged from 0.018 to >5000 nM (Figure 4). 31 Overall, median and geometric mean temsavir IC50s were 0.8 and 1.7 nM, respectively, and IC50s were <10 nM for 80% of isolates and >100 nM for 9% of isolates. IC50s were <10 nM for most subtype B isolates (84%), with 6% having IC50s >100 nM. Subtypes BF, F1, and BF1 had higher proportions (21%–38%) of isolates with IC50s >100 nM, and 5 of 5 (100%) CRF01_AE isolates had IC50s >100 nM.

Range of temsavir susceptibility observed within subtypes. Viruses with >5 isolates grouped by subtype. Horizontal solid lines indicate geometric mean. Symbols on dotted horizontal line had IC50s above the highest concentration tested.
In a genotypic analysis of 10,733 full-length gp160 sequences from all available isolates in the LANL HIV sequence database (through December 31, 2022), the most prevalent amino acids at relevant positions were the same as those seen in the subtype B consensus (S375, 72%; M426, 83%; M434, 88%; M475, 89%; Table 6). 30 The only amino acid polymorphism at these positions in ⩾10% of subtype B isolates was S375T (19% of isolates), which does not play a major role in determining temsavir susceptibility in vitro.14,28,30 S375H (99%) and M475I (77%) were predominant in subtype CRF01_AE. Among non-M HIV-1 groups, the consensus for group N isolates (N = 16) was 375M, 426L, and 434I, and 80 of 82 group O isolates carried S375H (in 10 cases in combination with M434I). 30 The impact of these amino acids on temsavir susceptibility in a group N or O background has not been confirmed; however, it seems likely that the efficacy of fostemsavir would be reduced in these viruses. The results from this analysis are consistent with other studies assessing the frequency of amino acid polymorphisms at positions 375, 426, 434, and 475 in clinical isolates of HIV-1.29,47–50 No major differences were reported in the distribution of RAPs or in vitro susceptibility to temsavir between isolates from individuals naive to ARVs or with prior ARV experience, or between CXCR4- and CCR5-tropic isolates.47–51

The top row for all amino acid positions shows the reference HXB2 sequence.
S375H/I/M/N/T/Y, M426L/P, M434I/K, M475I.
LANL database entries through December 31, 2022. Accessed August 23, 2024.
S375T has no measurable effect on in vitro temsavir susceptibility in an LAI *S375T background.
AA, amino acid; LANL, Los Alamos National Laboratory.
Clinical response to fostemsavir in study participants with subtype CRF01_AE
As described above, HIV-1 subtype CRF01_AE, which is predominant in Southeast Asia but uncommon elsewhere, shows considerably reduced susceptibility to temsavir in vitro. The RC of the BRIGHTE study included two participants with CRF01_AE virus.1,7 The baseline virus isolate for the first participant had a temsavir FC-IC50 of 298 along with a gp120 S375N substitution. This participant had HIV-1 RNA <40 copies/mL at Week 96 while receiving fostemsavir in combination with an OBT that included twice-daily dolutegravir. The baseline isolate for the second participant had a temsavir FC-IC50 of >4747 and gp120 S375H and M475I substitutions. This participant had an increase in HIV-1 RNA (0.473 log10 copies/mL) after 8 days of fostemsavir monotherapy but subsequently achieved HIV-1 RNA <40 copies/mL at Week 96 with fostemsavir plus OBT that included dolutegravir. It is not clear to what degree fostemsavir contributed to response in these individuals.
Discussion
The in vitro and clinical trial data described here are consistent in terms of (1) the wide ranges of in vitro temsavir susceptibility seen across clinical isolates of HIV-1, between different isolates of the same subtype, and between different individual clones from the same isolate9,10,14,31; (2) the occurrence of some cases of PDVF in the absence of detectable changes in in vitro temsavir susceptibility or emergent relevant amino acid substitutions2,27; (3) detectable treatment-emergent changes in in vitro temsavir susceptibility in the absence of detectable known RASs2,27; and (4) re-suppression after virologic failure in some participants who were HTE despite treatment-emergent genotypic and/or phenotypic evidence of reduced temsavir susceptibility.2,28 These observations highlight the complexity of accurately quantifying the role of gp120 mutations in cases of virologic failure during treatment with fostemsavir-based regimens in people with HIV-1 who are HTE.
Prior observations have shown that the impact of genotypic changes in gp120 on in vitro temsavir susceptibility is highly contextual, that different regions of the gp120 molecule interact to modify the temsavir binding pocket such that multiple amino acid substitutions play a role in determining susceptibility, 10 that variation in N-linked glycosylation sites could also impact temsavir susceptibility, 16 and that our current tools for measurement of the susceptibility of clinical isolates to temsavir do not capture the range of susceptibilities of the multiple different viruses present in the isolate. 14 Furthermore, binding studies show that temsavir can still inhibit the binding of CD4 to gp120 even in the presence of RAPs that have an impact on the in vitro temsavir susceptibility of pseudoviruses. 41 As a consequence of the large variability, limited clinical data, and context dependency of temsavir susceptibility, to date, no clinically relevant phenotypic cutoff or genotypic algorithms have been derived that reliably predict response to fostemsavir-based therapy in the HTE population; thus, pre-treatment resistance testing may currently offer limited value and is not stipulated in the product label.1,52 Given the marked reduction in susceptibility to temsavir among non-M HIV-1 groups and HIV-1 subtype CRF01_AE viruses, the European Medicines Agency product label advises against the use of fostemsavir to treat these strains, and the US label notes reduced in vitro antiviral activity.1,7 Due to the reduced susceptibility of CRF01_AE and the absence of an available resistance assay, the role of fostemsavir is limited in areas of the world where CRF01_AE is the prevalent strain, including Southeast Asia. For all other subtypes, there are currently no commercially available temsavir resistance tests, 23 and most online genotype interpretation tools do not provide updates that include the latest fostemsavir data.53–56 However, the HIV-GRADE online tool does provide an interpretation of env sequencing results based on an algorithm developed using multiple regularly updated data sources. 57 The current version of this interpretation may overestimate temsavir resistance and should be considered in the context of the potential activity of other ARVs that could be included in the regimen. In any case, guidelines encourage clinicians to consult with specialists in HIV drug resistance when interpreting genotypic test results and designing optimal ARV regimens. 23
The potential clinical benefits of fostemsavir therapy beyond virologic suppression should also be considered, particularly with regard to immunologic recovery. Guidelines recommend that, in the face of extensive ARV resistance, ongoing regimens should be designed to maintain CD4+ T-cell count, delay clinical progression, and minimize toxicity. 23 In BRIGHTE, fostemsavir plus OBT resulted in steady and substantial increases in CD4+ T-cell count and CD4+/CD8+ ratio through 240 weeks in both study cohorts, achieving levels that might not have been expected in participants with such advanced disease. Notably, the largest increases were seen among participants who were the most immunosuppressed at baseline, and substantial increases were seen in those who had Week 240 HIV-1 RNA ⩾40 copies/mL.2,5
Management of detectable virologic failure during treatment with fostemsavir will require a holistic approach, assessing not just viral load but also immunologic responses, pharmacokinetics, concomitant medications, tolerability, and resistance to other components in the regimen. In the absence of available options to construct a completely new regimen, there may be other changes that could be implemented, for example, improving treatment adherence or reducing drug–drug interactions. 23
Further analyses of long-term results and correlates of response in the ongoing BRIGHTE study, new data from the ongoing phase I/II SHIELD study (NCT04648280, fostemsavir in children and adolescents), and emerging data from real-world HTE cohorts 58 may yield further valuable information on the mechanisms and consequences of temsavir resistance and on the potential breadth of clinical benefits of fostemsavir.
Conclusion
The mechanisms of reduced susceptibility to temsavir are complex and highly context-dependent, currently precluding definitions of clinically relevant genotypic and phenotypic cutoffs. Despite this, many individuals show potent virologic response to fostemsavir-based regimens without baseline resistance testing, 5 and findings from clinical trials show that individuals who experience PDVF and emergent RASs can later re-suppress on fostemsavir.2,28 Furthermore, the unique mode of action of temsavir targeting gp120 before CD4 binding may provide clinical benefit beyond virologic suppression. Considerable and steady immunologic improvements through Week 240 were observed in the BRIGHTE study, even among individuals with viremia. 2 Thus, an individualized and holistic approach to the clinical management of people who are HTE and experience virologic failure during treatment with fostemsavir-based regimens is encouraged.