
Abstract
Background:
Cutaneous leishmaniasis (CL) ulcers exhibiting an inflammatory phenotype, characterized by purulent exudate, erythema, pain, and/or lymphatic involvement, are empirically treated with antibiotics.
Objective:
The spectrum of bacteria present in localized versus inflammatory phenotypes of CL is elucidated herein.
Methods:
Filter paper lesion impressions (FPLIs) from 39 patients with CL (19 inflammatory and 20 noninflammatory ulcers) were evaluated via real-time polymerase chain reaction (qPCR) and end-point PCR targeting: Staphylococcus aureus, Enterobacter cloacae, Streptococcus pyogenes, Enterococcus spp., Citrobacter freundii, Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, and 16S rDNA. Whole genome sequencing (WGS) was performed on six specimens.
Results:
In total, 30/39 (77%) patients’ ulcers had ⩾1 bacterium detected, which included the following species: S. aureus (n = 16, 41%), C. freundii (n = 13, 33%), P. aeruginosa (n = 12, 31%), E. cloacae (n = 12, 31%), K. pneumoniae (n = 11, 28%), Enterococcus spp. (n = 7, 18%), E. coli (n = 6, 15%), and S. pyogenes (n = 4, 10). Prevalence of bacterial species did not differ by CL phenotype (p = 0.63). However, patients with inflammatory phenotypes were, on average, over a decade older than patients with noninflammatory phenotypes (42 years vs 27 years) (p = 0.01). The inflammatory phenotype was more prevalent among ulcers of Leishmania Viannia braziliensis (58%) and L. V. panamensis (83%) compared to those of L. V. guyanensis (20%) (p = 0.0369).
Conclusion:
The distribution of flora did not differ between inflammatory and noninflammatory CL phenotypes. Further prospective analysis, including additional WGS studies of all CL ulcers for nonbacterial organisms, is necessary to determine the role of empiric antibiotic therapy in inflammatory and purulent CL.
Keywords
Introduction
Leishmaniasis is a vector-borne protozoan infection transmitted by female Phlebotomus spp. and Lutzomyia spp. sandflies. 1 While Leishmaniasis affects more than 12 million people worldwide, recent estimates show that approximately 350 million people in endemic countries across Northern Africa, the Middle East, Central and South America, and parts of Asia are at risk for contracting the disease. 2 Most causative species of Leishmania are classified according to one of three major clinical manifestations with which they are associated: cutaneous leishmaniasis (CL), visceral leishmaniasis, and mucocutaneous/mucosal leishmaniasis (MCL/ML). 2 The more than 20 infecting species of Leishmania are also divided between New World and Old World. 3 Specific to the New World is the Viannia subgenus, including Leishmania Viannia braziliensis, L. V. guyanensis, L. V. lainsoni, L. V. peruviana, and L. V. panamensis, with a propensity to cause MCL/ML. 3 Pathogenesis relies primarily on the interplay between the infecting Leishmania species and host immune responses. 2 Disease manifestation is often the result of the prevailing Th1 or Th2-biased response, involving a number of cytokines and chemokines differentially expressed as a result of infecting species and underlying host immune status. CL is often characterized by a Th1-biased response, associated with healing; whereby a mixed Th1/Th2 profile is exhibited in MCL/ML. 4
CL is the most common syndrome with an estimated 0.7–1.2 million cases worldwide. 1 CL is characterized by single or multiple ulcerating skin lesions typically localized at the site of the sandfly bite, but may also appear distally. 3 Ulcerative lesions are usually self-healing, but often persist for months or years if left untreated, with the risk of developing into ML approximately 1–5 years after initial healing of a CL lesion caused by primarily Viannia strains of Leishmania found in Latin America. 5 Moreover, specific strains of L. V. braziliensis, such as those containing genetic polymorphisms in parasite kinetoplast DNA minicircles and heat-shock protein 70, respectively, have been associated with treatment failure of CL and progression to MCL/ML.6,7 Clinical manifestations of CL can range from small noninflammatory lesions < 4 that may self-heal—often referred to as “localized CL”—to more complicated inflammatory ulcers characterized by pain, erythema, and purulent exudate with or without lymphatic involvement suggestive of bacterial co-infection, requiring additional therapy. 8 In addition, ulcerations of the skin in CL are associated with strong local pro-inflammatory responses to Leishmania-infected host macrophages and necrosis of the dermis, thus producing an ulcer either heavily inflamed, erythematous, or painful. 3
To date, causative species of Leishmania vary in their susceptibilities to different antimonial and other anti-leishmanial therapies. 9 As a result, there are CL treatment guidelines that recommend considerations for the size of the lesion, risk for developing ML, drug toxicities such as hepatotoxicity, cardiotoxicity, and nephrotoxicity, and prior treatment when prescribing CL therapy.8–13 Currently, there is a paucity of literature devoted to the management of patients presenting with the inflammatory phenotype, despite the anecdotal clinical practice of universal treatment of these ulcers with antibiotics prior to anti-leishmanial therapy. Although such inflammatory ulcers may appear to mimic a secondary bacterial infection, there is a scant evidence base supporting these common antimicrobial treatment practices.14–16
Antimicrobial stewardship and evidence-based management guidelines for inflammatory CL ulcers would benefit from knowledge of the microbial differences between inflammatory and noninflammatory CL ulcers in order to elucidate potential bacterial contribution to CL phenotype. Fontes et al. 15 demonstrated the colonization of various bacterial contaminants and pathogens in ulcers of American tegumentary leishmaniasis, prompting clinicians to consider bacterial contaminants in the treatment of CL ulcers. Additionally, detection of Enterobacterales such as Enterobacter cloacae, Klebsiella oxytoca Pseudomonas spp., and Staphylococcus spp.,14–16 and in particular, from the midgut of sandfly vectors, 17 has supported the notion that bacterial superinfection may be common. However, differences in clinical phenotype and how they might be influenced by the ulcer microbiome have been rarely considered. 15 Given the extensive knowledge surrounding common bacterial organisms as causative agents of skin and soft tissue infections (SSTIs), 18 we sought to document the spectrum of bacterial pathogens in inflammatory and noninflammatory ulcers of CL using polymerase chain reaction (PCR) and whole genome sequencing (WGS) to determine whether there were differences in bacterial spectra colonizing inflammatory compared to noninflammatory ulcers of CL.
Methodology
Specimen and clinical data collection
Surplus, discard filter paper lesion impressions (FPLIs) of CL ulcers submitted to the Public Health Ontario Laboratory (PHOL) between 2012 and 2018 were collected and stored at −20°C following clinical diagnostic testing. Additional prospectively collected FPLIs for patients enrolled in clinical diagnostic studies at the Leishmania Clinic of the Instituto de Medicina Tropical “Alexander von Humboldt,” Lima, Peru over the same time period were also enrolled following written informed consent. Research ethics board approval was obtained from Public Health Ontario (#2015-048.01 and 2017-052.01) and the Universidad Peruana Cayetano Heredia (#60401). De-identified clinical data for source patients collected from test requisitions and clinical charts were stratified into “inflammatory” and “noninflammatory” phenotypes as per the Infectious Diseases Society of America (IDSA) guidelines, 8 where an inflammatory phenotype was defined as an ulcer with associated erythema, purulent exudate, pain, and/or lymphatic involvement; whereas a noninflammatory phenotype was defined as localized CL (LCL) of <4 ulcers in number. 8
DNA extraction
DNA extraction for end-point PCR and real-time PCR (qPCR) was performed using the Qiagen DNA Mini Kit (Qiagen, Germantown, MD, USA) by soaking FPLIs in 250 µL of 1 × TE (10 mM Tris-HCl containing 1 mM EDTA·Na2) for 5 min prior to extraction. DNA was eluted with 60 μL of Buffer AE (10 mM Tris-Cl; 0.5 mM EDTA) and stored at −20°C prior to use. DNA extraction for WGS was performed using the ZymoBIOMICS DNA/RNA Miniprep Kit (Zymo Research, Tustin, CA, USA). Two sets of extractions were performed on each specimen where FPLIs were soaked in 500 µL of 1 × TE for 20 min prior to extraction. Deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) were extracted using the DNA & RNA Parallel Purification protocol with a 20-minute bead beating step. DNA was eluted with 50 µL DNase/RNase-free water and combined to maximize yield.
Leishmania species identification and confirmation
Leishmania genus 18S real-time PCR was performed as previously described. 19 Species identification included analysis of the internal transcribed spacer 1 (ITS1), ITS2, cysteine proteinase B (CPB), heat-shock protein 70 (HSP70), and mannose phosphate isomerase (MPI) by PCR, restriction fragment length polymorphism (RFLP) analysis, and Sanger sequencing.20–22 PCR–RFLP analysis of the ITS1 region can only differentiate L. V. braziliensis from the other species within the Viannia subgenus (L. V. guyanensis, L. V. peruviana, L. V. panamensis, L. V. lainsoni). Thus, PCR–RFLP and sequencing analysis of the CPB, HSP70, MPI, and ITS2 regions were required to differentiate species within the Leishmania Viannia subgenus complex and to provide a confirmation of the species identified in the initial ITS1 assay. Purified PCR product was used for Sanger sequencing as per Big Dye protocol (Life Technologies, Carlsbad, CA, USA). Sequenced products were purified and analyzed using the Applied Biosystems 3130xl Genetic Analyzer and Basic Local Alignment Search Tool (BLAST) searched for highest homology.
Detection of bacterial pathogens
Quantitative real-time PCR
16S rRNA qPCR along with the following six qPCR assays targeting Staphylococcus aureus, S. pyogenes, Escherichia/Shigella spp., Citrobacter freundii, Enterobacter/Klebsiella spp., and Enterococcus spp. (Qiagen, Cat# BBID00314AR, BBID00332AR, BBID00146AR, BBID00108AR, BBID00139AR, BBID00141AR, respectively) (Table 1) was performed on the Applied Biosystems 7900 Fast RT-PCR System using 5 µL of DNA as per manufacturer protocol. A specimen with a cycle threshold (Ct) value less than or equal to 40 was considered positive for the microbial-specific target. A pan 16S rDNA qPCR assay was also performed on the Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) using 12.5 µL Taqman Universal Master Mix and previously published primers and probes. The following conditions were used: 2 min at 50°C, 10 min at 95°C and 40 cycles of 15 s at 95°C and 1 min at 60°C (Table 1).
Primer and probe* sequences of bacterial targets.

Probe sequences for the species-specific qPCR assays are proprietary to Qiagen.
Fwd, Forward primer; qPCR, real-time polymerase chain reaction; Rev, Reverse primer, R*, A or G.
End-point PCR
The following bacterial targets were detected using end-point PCR assays performed on the Applied Biosystem Veriti® 96-well Thermal Cycler (Thermo Fisher Scientific): S. aureus, E. cloacae, S. pyogenes, Enterococcus spp., C. freundii, E. coli, P. aeruginosa, K. pneumoniae, and pan-bacterial 16S rDNA (Table 1). The annealing temperatures including primer and probe sequences varied among different microbial assays (Table 1). For each reaction, 3 μL of DNA was used in addition to 0.2 µL of Life Tech Native Taq, 2.5 µL of 10X Buffer, 0.5 µL of dNTP and 0.75 µL of MgCl2. The following conditions were used: 5 min at 94°C, 40 cycles of 45 s at 94°C, 30 s at 56°C and 1 min at 72°C, followed by 10 min at 72°C. A total of 5 μL of product was loaded onto 1% agarose gel and visualized for bands under UV.
Sanger sequencing
All sequencing reactions were performed on Applied Biosystems 3130xl Genetic Analyzers using the Big Dye v3.1 Cycle Sequencing Kit. The following cycling conditions were used: 1 min at 96°C, 25 cycles of 10 s at 96°C, 5 s at 50°C and 4 min at 60°C. PCR product was cleaned using 45 µL of SAM™ Solution (Applied Biosystems, Carlsbad, CA) and 10 µL of XTerminator beads on a vortex for 30 min. The product was centrifuged for 2 min at 2000 g prior to being loaded. Data were standardized using the Sequencing Analyzer program, Vector NTI® software (ThermoFisher Scientific, Carlsbad, CA) was used to assemble the sequences and BLAST search engine on the National Centre for Biotechnology Information database was used to analyze the sequences with highest homology.
WGS and bioinformatics analysis
In a proof-of-concept sub-analysis, a selection of six samples were analyzed in parallel using WGS. Genomic DNA was submitted to The Hospital for Sick Children—The Centre for Applied Genomics facility in Toronto, Canada for genomic library preparation and sequencing as a fee-for-service. DNA samples were quantified using Qubit High Sensitivity Assay Qubit dsDNA HS Assay Kits (ThermoFisher Scientific, Carlsbad, CA, USA), and sample purity was checked using Nanodrop OD260/280 ratio. Two-hundred ng of DNA was used as input material for library preparation using the Illumina TruSeq Nano DNA Library Prep Kit following the manufacturer’s recommended protocol. In brief, DNA was fragmented to 450 bp on average using sonication on a Covaris LE220 instrument; fragmented DNA was end-repaired, A-tailed and indexed TruSeq Illumina adapters with overhang-T were added to the DNA; adapter-ligated DNA was enriched by PCR under the following conditions: initial denaturation at 95°C for 3 min, followed by six cycles at 98°C for 20 s, 60°C for 15 s and 72°C for 30 s, and a final extension step at 72°C for 5 min and hold at 4°C. Libraries were validated on a Bioanalyzer DNA High Sensitivity chip to check for size and absence of primer dimers, and quantified by qPCR using KAPA Library Quantification Illumina/ABI Prism Kit protocol (KAPA Biosystems, Wilmington, MA). Validated libraries were pooled in equimolar quantities and paired-end sequenced on one lane on the Illumina HiSeq 2500 platform following Illumina’s recommended protocol to generate paired-end reads of 125-bases in length using a high output flow cell with the V4 chemistry. Raw reads generated from WGS were analyzed by CosmosID (Rockville, MD).
Statistical analysis
Descriptive statistics—including mean with standard deviation, median with ranges, and calculated proportions—were performed on continuous (age) and categorical (e.g., sex and clinical phenotype) variables, respectively. Comparative statistics on categorical variables were analyzed using Fisher’s exact test, chi-square, and chi-square test for trend (Cochran-Armitage test for trend); while continuous variables were compared by Mann–Whitney U-test. All statistical analyses were calculated via GraphPad Prism Version 6.01 (La Jolla, CA, USA) and MedCalc (MedCalc Software Ltd, Belgium). Significance was set at p < 0.05.
Results
Clinical and diagnostic data
In total, lesions from 39 patients were examined: 9 (23%) from the PHOL, 12 (31%) from Hospital Cayetano Heredia in Lima Peru, and 18 (46%) field isolate from Pichanaki, Peru—all of whom traveled from/currently reside in a CL endemic country. Nineteen (49%) ulcers fulfilled the criteria for an inflammatory phenotype, whereas the remaining 20 (51%) of ulcers had a noninflammatory phenotype (Table 2). Median age of all patients was 31 years (8–72 years), however, patients with the inflammatory phenotype were, on average, over a decade older than patients with the noninflammatory phenotype (42 years vs 26.5 years) (p = 0.01). No differences related to sex were observed, where 12 (63.2%) lesions in the inflammatory group occurred in males versus 7 (35%) lesions in the noninflammatory group occurred in males (p = 0.32) (Table 2).
Clinical and demographic correlates of 39 patients with ulcers due to CL stratified by phenotype.
Comparison of male and female.
CL, Cutaneous leishmaniasis.
Causative species
Species identification revealed the following prevalence: 12 (31%) of 39 patients had lesions due to L. V. braziliensis, 10 (26%), 6 (15%), 2 (5%), 3 (8%), and 6 (15%) were due to L. V. guyanensis, L. V. panamensis, L. V. peruviana, L. V. lainsoni and unknown/unable to confirm species, respectively (Table 2). Species identification revealed no difference among clinical phenotypes as follows: 7 (37%) inflammatory ulcers and 5 (25%) noninflammatory ulcers were due to L. V. braziliensis compared to non-L. V. braziliensis (p = 0.50), 2 (10%) inflammatory ulcers and 8 (40%) noninflammatory ulcers were due to L. V. guyanensis (p = 0.06), 5 (26%) inflammatory and 1 (5%) noninflammatory ulcers were due to L. V. panamensis (p = 0.09), 1 (5%) inflammatory and 1 (5%) noninflammatory ulcers were due to L. V. peruviana (p = 0.66), 2 (11%) inflammatory and 1 (5%) noninflammatory ulcers were due to L. V. lainsoni (p = 0.66), 2 (11%) inflammatory ulcers and 4 (20%) noninflammatory ulcers were due to hybrid/unidentified species (p = 0.66) (Table 2 and Figure 1). However, compared to both L. V. braziliensis and L. V. panamensis, L. V. guyanensis ulcers were less likely to cause an inflammatory phenotype: 58% of ulcers due to L. V. braziliensis, 83% of ulcers due to L. V. panamensis, and 20% of ulcers due to L. V. guyanensis were inflammatory (p = 0.0369).

Distribution of causative Leishmania spp. across the inflammatory and noninflammatory phenotypes of CL.
Bacterial organisms
Of the 39 ulcers, 30 (77%; 15 inflammatory and 15 noninflammatory) had at least one bacterial species detected while 9 (23%, four inflammatory and five noninflammatory) had no bacterial species detected (p > 0.99) (Table 3). The following organisms were identified in ulcers using non-WGS methods: P. aeruginosa was detected in 12 (30%) of 39 ulcers; while S. aureus was detected in 16 (41%), C. freundii was detected in 13 (33%), E. cloacae was detected in 12 (31%), K. pneumoniae was detected in 11 (28%), Enterococcus spp. was detected in 7 (18%), E. coli was detected in 6 (15%), S. pyogenes was detected in 4 (10%), S. oralis was detected in 2 (5%), and S. epidermidis, S. thermophilus, Solobacterium spp., and Pelomonas puraquae were detected in 1 (3%) each, respectively (Table 3). There was no difference observed in the presence of the following organisms across phenotypes: P. aeruginosa was detected in 7 (37%) inflammatory versus 5 (25%) noninflammatory ulcers (p = 0.50); S. aureus was detected in 8 (42%) inflammatory versus 8 (40%) noninflammatory ulcers (p > 0.99); C. freundii was detected in 6 (32%) inflammatory versus 7 (35%) noninflammatory ulcers (p > 0.99); S. pyogenes was detected in 3 (16%) inflammatory versus 1 (5%) noninflammatory ulcers (p = 0.34); E. cloacae was detected in 4 (21%) inflammatory versus 8 (40%) noninflammatory ulcers (p = 0.30); K. pneumoniae was detected in 5 (26%) inflammatory versus 6 (30%) noninflammatory ulcers (p > 0.99); E. coli was detected in 1 (5%) inflammatory versus 5 (25%) noninflammatory ulcers (p = 0.18); and Enterococcus spp. was detected in 4 (21%) inflammatory versus 3 (15%) noninflammatory ulcers (p = 0.69) (Table 3 and Figure 2). There was no difference in the number of ulcers containing pathogenic (S. aureus, S. pyogenes, P. aeruginosa, C. freundii, Enterobacter/Klebsiella spp., E. coli) versus opportunistic (S. epidermidis, S. thermophilus, Enterococcus spp., S. oralis, Solobacterium spp., P. puruqae) bacterial organisms observed across inflammatory and noninflammatory phenotypes of CL (p > 0.99) (Figure 3).
Spectrum of bacterial pathogens among inflammatory and noninflammatory CL.
Grouped “0,” “1,” “2,” and “3,” “4+” for comparison.
Compared the respective bacteria to the other bacterial species.
CL, Cutaneous leishmaniasis.

Distribution of bacterial organisms among patients with the inflammatory and noninflammatory phenotypes of CL.
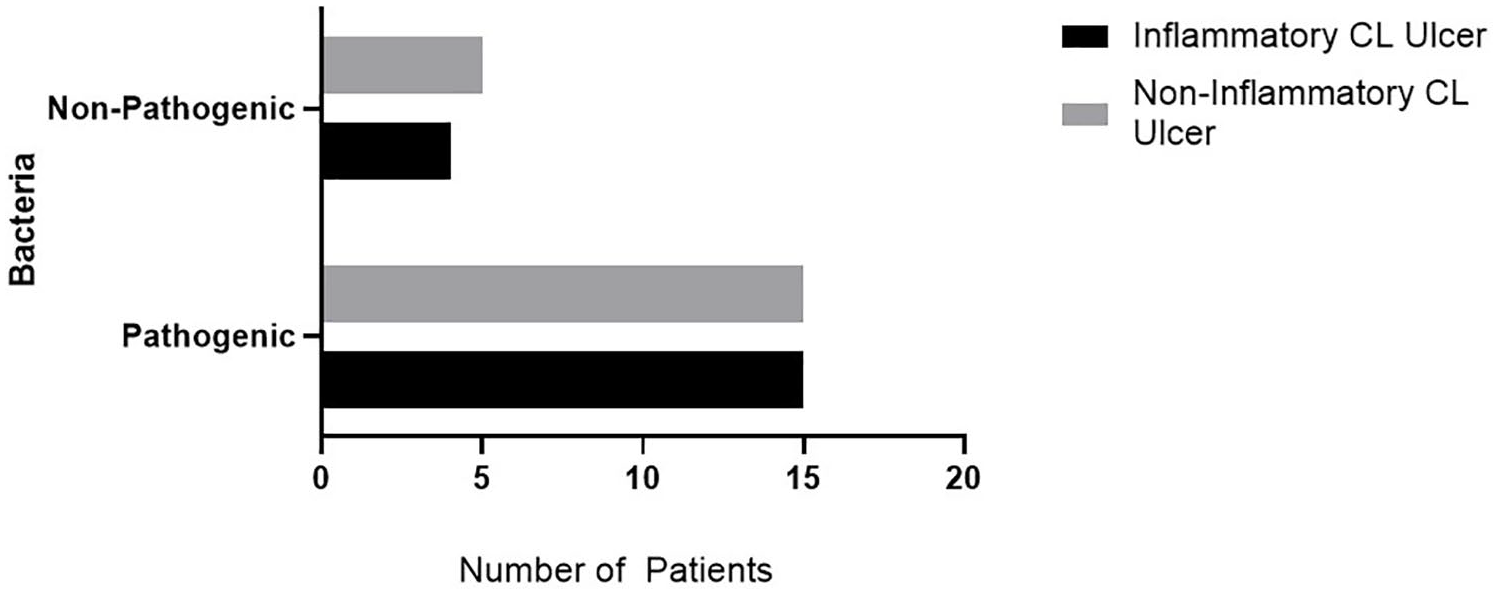
Distribution of pathogenic (S. aureus, S. pyogenes, P. aeruginosa, C. freundii, Enterobacter/Klebsiella spp.) versus nonpathogenic (S. epidermidis, S. thermophilus, Enterococcus spp.) bacteria among ulcers of inflammatory and noninflammatory CL.
WGS analysis
Of six ulcer FPLIs sent for WGS, 4 (68%) lesions were caused by L. V. braziliensis, 1 (16%) was due to L. V. peruviana, and 1 (16%) was below detection limits for Leishmania species identification; only four had sufficient sequence data for further WGS analysis. Among the four FPLIs with sufficient sequence data, WGS yielded identification of many environmental bacterial contaminants, with Brevundimonas nasdae, a gram-negative opportunistic pathogen, identified in all four samples. Other viral, parasitic, and fungal organisms were identified, including Melampsora pinitorqua, Staphylococcus phage, and Toxoplasma gondii. WGS and conventional PCR identified S. aureus and C. freundii concordantly. WGS remained more sensitive for P. aeruginosa, S. pyogenes, E. coli, and Enterococcus spp., however, remained less sensitive for Enterobacter spp. and K. pneumoniae (Table 4).
Comparison of WGS to conventional PCR methods for pathogen detection in subset of six samples.

NA, not applicable; PCR, polymerase chain reaction; WGS, whole genome sequencing.
Discussion
The present study demonstrates that detection of potentially pathogenic and nonpathogenic bacteria is common in ulcers of CL, regardless of phenotype, with bacterial species detected in approximately 77% of CL ulcers in this study. Although the frequency of CL ulcers harboring no detectable bacteria by targeted molecular assays was only 23%, this may be a result of our limited set of microbial assays, few samples with available WGS data, as well as low relative abundance and possibly restricted bacterial diversity that has been previously documented in CL ulcers. 32 Overall, we reported equal distributions of detectable organisms, pathogens, and nonpathogens among inflammatory and noninflammatory ulcers of CL. Furthermore, prevalence of bacterial species did not differ by CL phenotype. This finding suggests a minimal contribution of detected bacterial organisms to the pathogenesis of the severe inflammatory CL phenotype. Patients with the inflammatory phenotype were, on average, 15 years older than patients with the noninflammatory phenotype, which may have influenced ulcer bacterial microbiome due to age-related immune dysfunction (immunosenescence), however, this study was not designed to test such a hypothesis.
Case reports and studies have emerged examining the impact of secondary bacterial infections and healing times of patients with CL ulcers,14,33 which have shown conflicting results. High proportions of S. aureus present in the CL ulcers in our study were not particularly surprising, as this species is known to frequently inhabit many regions of the skin—exerting pathogenesis when it infects deeper tissues, the blood, or expresses pathogenic endotoxins or exotoxins. 34 The proportions of P. aeruginosa were descriptively different among the two groups, with P. aeruginosa detected in 44% of inflammatory CL ulcers versus 18% of all noninflammatory CL ulcers, however, this was not statistically significant. This relatively higher frequency of P. aeruginosa in inflammatory ulcers may suggest its potential association with the severe inflammatory phenotype given its highly pathogenic nature and propensity for multi-drug resistance, though the sample size in this study was too small to detect this difference. Based on the prevalence of P. aeruginosa, a larger sample size of 50 (power of 0.8 and significance of 0.05) in each of the respective inflammatory and noninflammatory ulcer arms would be required to detect a significant difference in pseudomonal prevalence across phenotypes. Previous studies have also reported Staphylococcus spp., Streptococcus spp., Enterococcus spp., Pseudomonas spp., and other opportunistic bacteria in LCL lesions that are free of signs of secondary infection, again supporting a lack of bacterial causality to the inflammatory phenotype. 15
Previous clinical reports have suggested earlier and prompt antibiotic therapy in cases where P. aeruginosa is present in cutaneous lesions in order to prevent severe mutilation, such as those seen in inflammatory CL ulcers. 35 It is known that P. aeruginosa has the intrinsic potential to develop antibiotic resistance through a variety of different mechanisms, causing pathogenesis in immunocompromised or highly comorbid individuals who are immunocompromised.36,37 We did note an association between age and disease severity, whereby older patients were more likely to manifest the inflammatory phenotype, yet we did not find a difference in bacterial spectra that may explain this observation.
Strengths of the current study include the use of both bacterial-specific qPCR and pan-bacterial end-point PCR assays, as well as WGS on a subset of samples, which allowed for proof-of-concept of improved sensitivity and specificity for detecting bacterial organisms relative to culture-based methodology carried out by other studies evaluating the microbiome of CL ulcers.15,38 Both WGS and PCR produced concordant results for the detection of S. aureus and C. freundii, however, depending on the organism, WGS or PCR would outperform the other. Culture-based methodology can largely exclude bacteria of low relative abundance and fastidious microorganisms, which may result in the under-representation of species present in the CL microbiome.
Limitations of this study include the small number of ulcers in each group, with varying causative Leishmania spp. We found no association between causative Leishmania species and CL phenotype, but whether or not this represents a true absence of association or was a result of a small sample size cannot be determined. Consequently, beyond WGS, we had no molecular corroboration of nonbacterial members of the ulcer microbiome, nor could we infer a colonization role versus infection for any detected organisms. Another limitation is our inability to detect the production of virulence factors or specific resistance genes in any of the organisms detected as such an objective was beyond the scope of this study. However, understanding which detected organisms are elaborating virulence factors within the ulcer microbiome is another dimension potentially underpinning causality that should be investigated in a prospective fashion in a larger clinical study.
Finally, ulcer chronicity and previous use of topical therapeutics are potential modifiers of representative organisms, such as use of antimicrobial ointments or oral cephalexin reducing burden of S. aureus and S. epidemmidis. 39 Comorbidities including diabetes and vascular disease would also theoretically contribute to ulcer microbiome, as would smoking and micronutrient status due to the likely synergistic effects of reduced vascular perfusion of tissue, impaired wound healing, dysfunction of adipose tissue leading to insulin resistance and consequent immunological changes, although we have no access to such data in this cohort. 40 Moreover, genetic polymorphisms of human leukocyte antigen (HLA) genes have been found to confer protection against or impart susceptibility to leishmaniasis.41,42 Again, these potential modifiers could be investigated prospectively in a larger clinical study and a thorough examination of both host- and parasite-specific factors.
Beyond the role of targeted antimicrobial therapies for secondarily infected CL ulcers, phytochemicals—that is, biologically active compounds found in plants—such as capsaicin, may also have a concomitant role in anti-leishmanial and anti-bacterial action. 43 Despite the very low quality of existing observational studies, ethnopharmaceutical compounds used in traditional medicine as well as novel compounds such as garlic, garad, neem, Mat Lippia, Physalis minima, and Morinda citrifolia were evaluated on humans with some level of cure achieved in participants of these studies, with comparable efficacy to current anti-leishmanial therapeutics.44–47 Future evaluation of such plant-based compounds in the context of secondarily infected ulcers may prove to be an all-encompassing therapeutic modality to combat both bacterial and leishmanial organisms and improve wound healing.
The overall representation of pathogenic and nonpathogen bacterial organisms was not associated with the inflammatory phenotype, however, P. aeruginosa may be present in higher frequencies in inflammatory compared to noninflammatory ulcers. Future studies with a larger sample size to study this association are warranted. As well, further examination of additional ulcers using WGS will clarify the bacterial composition between the two ulcer phenotypes, which may have implications for our understanding of antimicrobial stewardship in ulcerative leishmaniasis. The identification of other viral, fungal, and parasitic organisms raises the question of nonbacterial agents contributing not just to the ulcer microbiome but to the inflammatory phenotype, in particular. Such organisms could include the endosymbiotic Leishmania RNA Virus-1, 48 and may enhance parasitic virulence and drive host immune responses, a tantalizing explanation of older patients in this cohort developing the more inflammatory phenotype. Future work should focus on disentangling the interrelationships between host, parasite, and ulcer microbiome in order to improve clinical response, direct wound care, and better target antimicrobial therapy according to principles of stewardship.
Conclusion
This study demonstrated that the bacterial microbiology of the severe inflammatory phenotype of CL ulcers was not significantly different compared to that of the noninflammatory phenotype. Considering the alarming spread of antimicrobial resistance, our findings may inform clinical practice as antimicrobial therapy may not truly impact resolution of inflammatory CL ulcers. Rather, the diminution of the inflammatory appearance may simply be predicated upon adequate wound care and timely anti-leishmanial therapy. Future studies should include substantiating the current findings with larger sample sizes and focusing on other contributions to the pathogenesis of CL ulcer phenotypes to further inform management guidelines. The use of WGS may be warranted to assess bacterial and nonbacterial contributions to the CL phenotype as observed in this study, where other viral, fungal and parasitic organisms not captured from the outputs of singleplex assays can be identified. 49