
Abstract
Objectives
In order to study the phenotype-genotype relationship and to better understand the early consequences of the mutation, we would report the spectrum of electrocardiographic and genetic features in the relatives of hypertrophic cardiomyopathy (HCM) patients.
Methods
Participants underwent a comprehensive clinical assessment, electrocardiography, standardized and echocardiography and genetic testing. In probands, next-generation sequencing was performed using the gene panel associated with HCM, while in relatives, Sanger sequencing was used to screen for mutations identified in their individual probands.
Results
A total of 84 participants were included in this study. The interventricular septal and posterior wall thickness was highest in the G+/LVH+ group, followed by the G+/LVH− group, and was lowest in G−/LVH− group. Compared to the normal control group, the pathologic Q wave was statistically more prevalent in the G+/LVH− group. The prevalence of repolarization abnormalities and major abnormalities was highest in the G+/LVH+ group, followed by the G+/LVH− group, and lowest in G−/LVH− group.
Conclusion
Our results suggested that sarcomere mutations have early consequences on myocardial biology. These findings suggest the possibility of implementing a mutation carrier detection model within families affected by HCM, where ECG could play a central role when combined with other relevant clinical factors. Longitudinal studies on a cohort of G+/LVH− patients are required.
Keywords
Introduction
Hypertrophic cardiomyopathy (HCM) is an inherited cardiovascular heart disease associated with genetic mutations which encode for components of the heart's contractile system. 1 HCM has a global prevalence of approximately 0.2%, with a typical onset in the third decade of life; however, the symptoms can also manifest in newborn as well as elderly patients.2,3 HCM is characterized by left ventricular hypertrophy (LVH), which causes left ventricular outflow obstruction, diastolic dysfunction, myocardial ischemia, and mitral regurgitation. These structural and functional defects could result in a wide variety of symptoms, including fatigue, dyspnea, chest pain, palpitations, syncope, and even sudden cardiac death can be the first manifestation of the disease.4,5
However, LVH is not unique to HCM and does not encompass the entire disease spectrum. Regardless of the presence of a pathogenic mutation, wall thickness in early childhood is typically normal and can persist for decades.6,7 In addition, this manifestation could be resembled by other conditions including infiltrative and metabolic cardiomyopathies, hypertension, and athletic remodeling. 8 The delayed penetrance and poor specificity of this key diagnostic feature make it difficult to identify family members who are at risk of developing HCM. Genetic tests could provide definitive identification of such individuals’ relatives who have inherited a pathogenic sarcomere mutation (G+) but do not yet have the diagnostic clinical features of HCM (G+/LVH−). 9 Additionally, the unavailability and high cost of genetic tests could limit their use in routine clinical practice, as their widespread and consistent application is regarded as impractical.
In contrast to genetic testing, the electrocardiogram (ECG) is a noninvasive, low-cost, high-availability test that is routinely used in the evaluation of patients with HCM.10,11 Mutation carriers who have yet to develop LVH may have altered myocardial damage at a microscopic level, which is indirectly expressed by certain electrocardiographic features that can be observed during screening, such as LVH, pathological Q-wave and ST-T abnormalities. 11 Therefore, these features have been proposed to be an earlier and more sensitive manifestation of sarcomere mutations than increased LV wall thickness.12,13 In this sense, ECG abnormalities may serve as cost-effective, easily accessible noninvasive indicators to identify individuals suitable for genetic testing in order to establish a conclusive diagnosis of HCM. Additionally, they can facilitate regular monitoring to detect the emergence of clinical symptoms associated with LVH. 6 Recognizing this potential, we conducted this study with the primary aim of thoroughly describing the electrocardiographic and genetic features in the relatives of HCM patients. Our study's results could serve two purposes: first, ECG abnormalities in individual mutation carriers could provide additional insights into the early impact of mutations in the development of subclinical phenotypes, thereby providing further knowledge about the genotype–phenotype relationship. Second, differences in ECG abnormalities among patient groups could suggest the possibility of implementing a mutation carrier detection model within families affected by HCM, where ECG could play a central role when combined with other relevant clinical factors, particularly in resource-limited settings, such as developing countries and primary settings, where genetic testing may not be readily available.
Methods
Study design
A cross-sectional study was conducted from August 2021 to August 2022 at the Vietnam National Heart Institute on 28 HCM probands and 56 of their relatives.
Patient eligibility
In the proband group, eligible participants were (1) those diagnosed with HCM according to the American Heart Association (AHA) 2020 criteria (echocardiographic evidence of hypertrophy, defined as maximal LV wall thickness ≥15 mm or z score > 2.5 in participants <18 years of age 14 ) and mutations classified as pathogenic/likely pathogenic/uncertain significance according to ACMG 2015 criteria. 15
In relative groups, eligible participants were first-degree relatives (including proband's parents, siblings, and children). Based on the results of 2D echocardiography and genetic testing, relatives will be classified into one of the following groups: (1) relatives with a mutation and echocardiographic evidence of LVH (G+/LVH+), applying similar criteria as probands but with a lower threshold for LVH in echocardiography (maximal LV wall thickness ≥13 mm or z-score > 2 in participants <18 years of age 14 ); (2) mutation carriers without echocardiographic evidence of LVH (G+/LVH−); and (3) related normal controls (G−/LVH−).
Exclusion criteria included: hypertension (systolic blood pressure >140 mmHg or diastolic blood pressure >90 mm Hg or receiving treatment), coronary artery disease, greater than mild intrinsic valvular heart disease, congenital heart disease, prior invasive septal reduction, severe electrolyte imbalances (Na+ below 125 mEq/L or above 155 mEq/L; K+ below 2.5 mEq/L or above 6.0 mEq/L), heart failure, pericarditis, myocarditis, channelopathies, and hyperthyroidism/hypothyroidism.
Data collections
Probands were clinically examined at baseline, with evaluations including physical examination, medical history, electrocardiography, standardized echocardiography, and genetic testing.
If the patients meet the inclusion criteria, we would call their first relatives to explain the study objectives. If they agree to participate, these relatives will be scheduled for an appointment at the study site, where they undergo a comprehensive clinical assessment, ECG, echocardiogram and blood tests similar to the probands. Genetic testing was performed using Sanger sequencing in the relatives. Figure 1 summarizes the screening and recruitment process of this study.

Study flow chart.
Clinical features
The following clinical features were obtained at study inclusion: (1) gender; (2) age at diagnosis; (3) systolic/diastolic blood pressure; (4) heart rate; (5) dyspnea degree according to the New York Heart Association classification; and (6) angina degree according to the Canadian Cardiovascular Society classification. All the information has been recorded in detail in the patient's medical charts.
Electrocardiographic features
12 lead ECGs were recorded by a healthcare worker who has been appropriately trained in this procedure, with a speed of 25 mm/s. The ECGs were recorded at the time of administration when the patient had rested for about 10–15 min. The task of ECG analysis was delegated to a board-certified cardiologist who was blind to clinical, genetic, and echocardiographic data. Heart rate, QRS axis, QRS, and QT intervals were measured according to the standard criteria. 16 The QT interval was corrected for the heart rate according to Bazett's formula (QTc = QT/√RR).
Bundle branch block, interventricular conduction delay, and atrioventricular block were defined according to the standard AHA criteria, as previously reported. 17
Pathologic Q wave was defined when it appeared in at least two consecutive leads and met both the abnormality criteria for amplitude (≥1/3 of R wave height or >0.3 mV) and duration (≥30 ms). 16 It is important to note that in this study, the term “pathologic Q wave” was not used to refer to Q waves associated with myocardial infarction, which exhibit distinct characteristics as discussed elsewhere. 18 T-wave inversion was defined as ≥0.1 mV in ≥2 contiguous leads. 19 If ST-segment depression was present in at least two adjacent leads with a depth of >0.1 mV (for upsloping) or >0.05 mV (for horizontal or downsloping), it was considered pathological. 20 If there were abnormalities in the ST-segment or T waves but they didn’t match the criteria for T-wave inversion or ST-segment depression, they were regarded as nonspecific ST or T-wave abnormalities.
Standard criteria were employed to define LVH, including Romhilt-Estes (where LVH was defined as a score of ≥4), 21 Cornell (where LVH was defined as Sv3 + RaVL ≥ 2.1 mV in women or ≥2.9 mV in men), 22 Sokolow-Lyon (where LVH was defined as SV1 + RV5 or SV1 + RV6 ≥ 3.5 mV), 23 and total voltage (where LVH was defined as the sum of the greatest positive and negative QRS deflection in 12 standard leads >17.5 mV). 24 ST-segment depression, T-wave inversion, Q waves, or any of the LVH criteria were considered to be major ECG abnormalities, as presented in Figure 2. In the absence of major abnormalities, minor ECG abnormalities included nonspecific ST/T abnormalities, left atrial abnormality, or delayed interventricular conduction (QRS > 110 ms).

Major electrocardiogram abnormalities.
Echocardiographic features
Echocardiography was performed by an independent cardiologist who was blind to the clinical, genetic, and electrocardiographic data. The following parameters would be assessed: eject fraction calculated by Simpson's method, interventricular septal diastolic, interventricular septic systolic, left ventricular end-diastolic dimension (Dd), left ventricular end-systolic dimension (Ds).
Genetic testing
Genomic DNA was extracted from the peripheral blood lymphocytes using the DNA blood mini kit. All gDNA samples were of a high quality with a DNA concentration >20 ng/µL and an OD 260/280 from 1.8 to 2.0. In 28 probands, next generation sequencing (NGS) was performed using the gene panel associated with HCM, including ACTC1, MYBPC3, MYH6, MYH7, MYL2, MYL3, TNNC1, TNNI3, TNNT2, TPM1, ACTN2, CASQ2, CSRP3, FHL1, LDB3, NEXN, TCAP, TTN, VCL, JPH2, GLA, LAMP2, and TTR. In 56 relatives, Sanger sequencing was used to screen for mutations identified in their individual probands.
The identified variants were then classified as “pathogenic (P),” “likely pathogenic (LP),” “uncertain significance (VUS),” “likely benign (LB),” or “benign (B)” by the Clinvar database and according to the 2015 ACMG/ACP guidelines. 15
Statistical analysis
We described qualitative variables as frequencies and percentages and quantitative variables as means (standard deviation, SD) or medians (interquartile range, IQR). Continuous variables with a normal distribution were compared using a t-test or one-way ANOVA, whereas continuous variables with an abnormal distribution were compared using the Mann-Whitney test. To compare categorical variables, the chi-square test or Fisher exact test was employed. A p-value <.05 was considered statistically significant for the comparison of two groups, while a p-value <.017 (.05 divided by three comparison groups) was considered statistically significant to apply a post hoc Bonferroni correction for multiple comparisons across the three status groups. Stata/BE 17 (StataCorp LLC, College Station, TX, US) was used for all analyses.
Ethical consideration
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Hanoi Medical University under decision No. 3135/QĐ-ĐHYHN dated July 22nd, 2021. Informed consent was obtained from adult patients, and parental consent/youth assent was obtained for younger participants before participating in the study. The investigators were responsible for protecting the privacy and confidentiality of patients as per Vietnam's regulations and Good Clinical Practice.
Results
A total of 84 participants from 28 families were included in this study, of which, 31 were overt HCM (G+/LVH+), 20 were mutation carriers without echocardiographic evidence of LVH (G+/LVH−) and 33 were healthy controls (G−/LVH−). The mean age and prevalence of syncope were higher in the G+/LVH+ group. Although minor differences in heart rate and blood pressure were noted among the status groups, all were within the normal ranges. The interventricular septal and posterior wall thickness was highest in the G+/LVH+ group, followed by the G+/LVH− group, and was lowest in the G−/LVH− group. Clinical and echocardiographic features of the participants are summarized in Table 1.
Clinical/echocardiographic features and genetic backgrounds of the participants (n = 84).
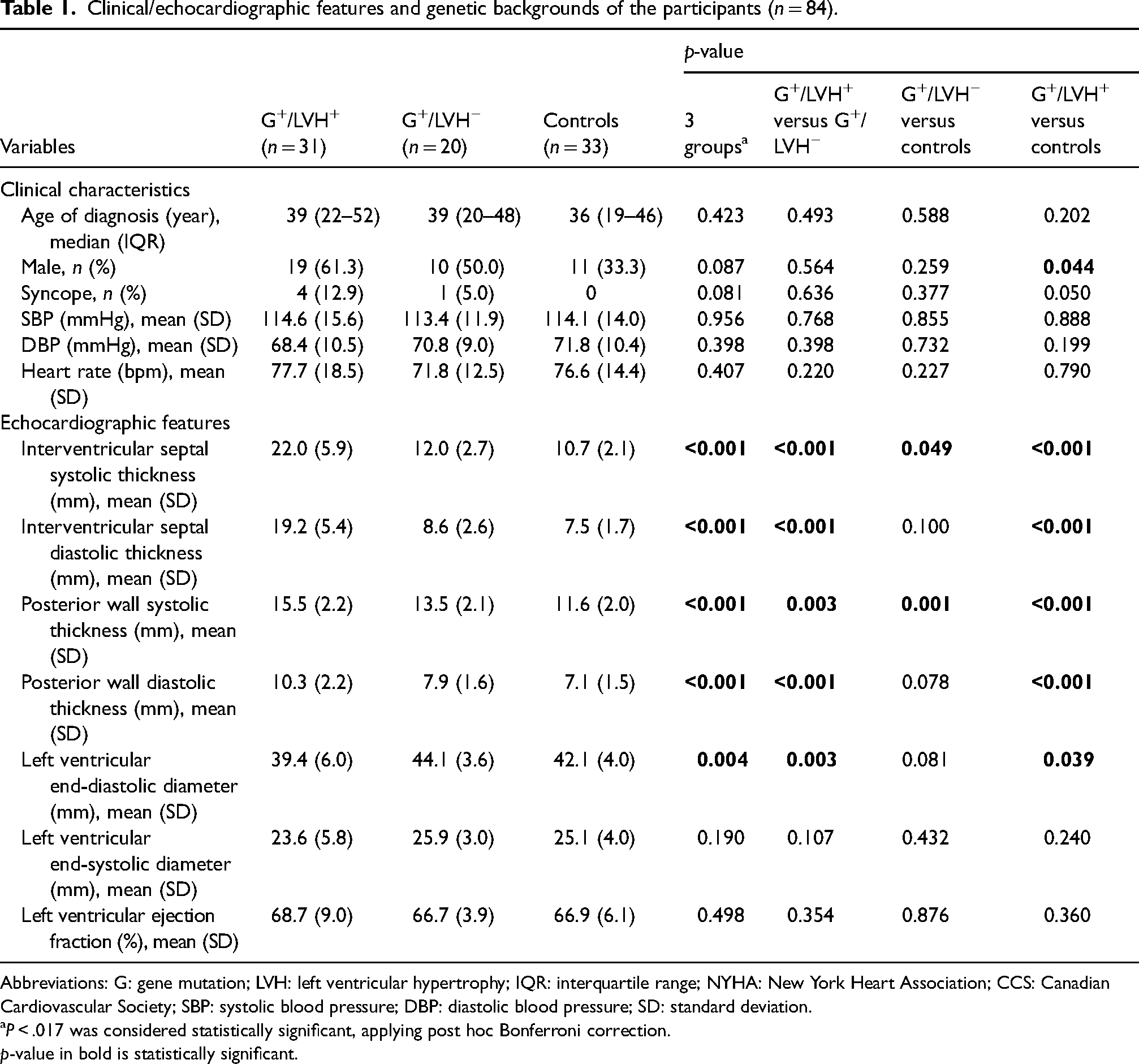
Abbreviations: G: gene mutation; LVH: left ventricular hypertrophy; IQR: interquartile range; NYHA: New York Heart Association; CCS: Canadian Cardiovascular Society; SBP: systolic blood pressure; DBP: diastolic blood pressure; SD: standard deviation.
aP < .017 was considered statistically significant, applying post hoc Bonferroni correction.
p-value in bold is statistically significant.
In 28 probands underwent genetic testing, the NGS identified 28 different variants. According to the ACMG, 21 (75%) variants were classified as pathogenic, three (10.7%) variants were classified as likely pathogenic and 4 (14.3%) variants were classified as variants of uncertain significance. The majority of the variants were missense (78.6%) and nonsense (17.8%). There was only one frameshift variant, which was a deletion in exon 17 of the MYH6 gene. No novel mutations were detected in this study. Based on the mutations identified in probands, we screened a total of 56 relatives and detected those mutations in 41.1% of the relatives (23/56 relatives). The pathogenic variants identified in this study are described in Table 2.
The pathogenic variants identified in HCM probands and relatives.

Abbreviations: dbSNP: database for single nucleotide polymorphisms; ExAC: exome aggregation consortium; gnomAD: the genome aggregation database; ACMG: American College of Medical Genetics and Genomics; pt: patient; NA: not available data; P: pathogenic; PM: evidence of pathogenicity moderate; PP: evidence of pathogenicity supporting; PS: evidence of pathogenicity strong; PVS: evidence of pathogenicity very strong; HCM: hypertrophic cardiomyopathy.
The heart rhythm and electrical axis were normal, and the conduction disturbance and atrioventricular block were absent in the majority of the cases. Compared to the normal control group, the pathologic Q wave was statistically more prevalent in the G+/LVH− group, while it was similar between the G+/LVH+ and G+/LVH− groups. The prevalence of repolarization abnormalities and major abnormalities was highest in the G+/LVH+ group, followed by the G+/LVH− group, and lowest in the G−/LVH− group, while the prevalence of LVH (according to standard criteria) was similar between G+/LVH− and normal control groups. The electrocardiographic characteristics of the participants are presented in Table 3. The stratification by the type of variant (P/LP vs VUS) showed no significant difference in ECG characteristics between stratified groups, as presented in Table 4.
Electrocardiographic features of the participants (n = 83). a

Abbreviations: G: gene mutation; LVH: left ventricular hypertrophy.
aOne patient was left out of the analysis due to the pacemaker rhythm.
bP < .017 was considered statistically significant, applying post hoc Bonferroni correction.
cStandard criteria were used to define LVH, including Romhilt-Estes, 21 Cornell, 22 Sokolow-Lyon, 23 and total voltage. 24
p-value in bold are statistically significant.
Electrocardiographic features based on mutation type (n = 83). a

Abbreviations: G: gene mutation; LVH: left ventricular hypertrophy.
aOne patient was left out of the analysis due to the pacemaker rhythm.
bChi-square test.
cFisher exact test.
dStandard criteria were used to define LVH, including Romhilt-Estes, 21 Cornell, 22 Sokolow-Lyon, 23 and total voltage. 24
p-value in bold is statistically significant.
Discussion
The most common ECG presentation among mutation carriers in this study was pathologic Q wave, which was more prevalent in the G+/LVH− group than in the normal control group but similar to the overt HC group. In a study with 20 HCM patients and 10 normal controls, intracoronary ECG revealed Q waves associated with regional wall-motion abnormalities in 12 HCM patients, whereas none of the control subjects displayed Q waves. 25 In a 2011 cross-sectional study conducted on 213 participants (57 G+/LVH+, 76 G+/LVH−, and 80 healthy controls), Q waves were a highly specific (98% specificity) marker for LVH mutation carriers, which presented in 25% of G+/LVH and 3% of controls (p < .001). However, in cardiac magnetic resonance (CMR), myocardial scar or perfusion abnormalities were not presented. 26 This finding suggests that, unlike in ischemic heart disease, Q wave in HCM patients is not likely due to the myocardial fibrotic scar. In previous studies evaluating the correlation between EKG and CMR, pathological Q waves were thought to be a result of potential imbalances, in which the initial QRS vector is altered due to increased electrical forces caused by disproportionate hypertrophy of the basal septal and/or ventricular free wall, which are not countered by apical forces.27,28 However, other studies attempting to clarify the anatomic basis of Q waves in HCM through CMR imaging have yielded conflicting explanations.29,30
In this study, abnormal repolarization was also a characteristic ECG feature, with a decreasing prevalence in G+/LVH+, G+/LVH− and healthy controls. Previous studies have also reported a similar pattern, with the prevalence of repolarization abnormalities and major abnormalities highest in the G+/LVH+ group, followed by the G+/LVH− group, and lowest in the G−/LVH− group.26,31 The mechanism of abnormal repolarization in mutation carriers has not been understood, however, local myocardial fibrosis is considered to be involved. In a 2016 study conducted in 88 HCM patients, repolarization disturbances were correlated with late gadolinium enhancement scores in cardiac magnetic resonance. 27 This finding implied that myocardial disarray and fibrosis are likely to cause heterogeneity of the ventricular refractory period and intraventricular conduction, resulting in negative T waves and QT dispersion.
Our study did not show a difference between the prevalence of LVH determined by ECG criteria between the G+/LVH− group and the healthy control group. Although left ventricular wall thickness plays a key role in the diagnosis, the ECG criteria alone serve as a low sensitivity and specificity marker in mutation carriers, as reported in previous studies.26,28,32 The ECG criteria for LVH may be influenced by physiological conditions such as myocardial hypertrophy in athletes, or secondary to other conditions including hypertension, valvular disease, and coronary heart disease. False negative cases with increased left ventricular load but no increase in QRS complex amplitude (obesity, edema) or false positive cases with increased QRS amplitude but no increase in left ventricular load (thin chest wall, young men) can potentially confound the clinical diagnosis. 33 In a 2015 study which examined the diagnostic value of ECG-LVH using CMR as the gold standard, the results revealed a discrepancy in diagnostic performance, thus suggesting that LVH by ECG and imaging are likely to be two distinct phenotypes. 34 As recommended by recent guidelines for preparticipation screening using ECG,35,36 the accurate detection of LVH in HCM requires careful inspection for repolarization disturbances, QRS axis deviation and other associated abnormalities. Isolated LVH-ECG without accompanying repolarization changes is more likely to lead to poor diagnostic accuracy.
In our study, the prevalence of mutation in the patient's relatives was 41.1%, which was similar to previous studies.26,37 This conformed to the practically asymptomatic characteristics of the relative population. For relatives with a negative genetic test result, disease-free status could be assumed, relieving them of the psychological and financial burden of disease monitoring and physical activity restriction.14,38 In contrast, relatives with positive results without clinical phenotypic expression present challenges for cardiologists, since HCM could present after the age of 40. 39 Therefore, a single evaluation could not confirm whether these individuals have developed HCM symptoms and consequences. According to the AHA 2020 recommendation, this group should be systematically monitored in terms of clinical, ECG, and cardiac imaging modalities based on age and clinical status. 14
The major limitation of our study is the relatively small sample size, in addition to the fact that our analysis concentrated on narrow subgroups of patients, making it difficult to reach a definitive conclusion. Also, because HCM is fundamentally a disease that progresses over time, the cross-sectional nature of this study may not fully represent the phenotypic expression of the participant, as well as the genotype-phenotype association.
Conclusion
In this study, ECG features that are consistently present in genetic mutation carriers were identified. The presence of Q waves and repolarization abnormalities in mutation carriers with normal LV wall thickness suggested that sarcomere mutations have early consequences on myocardial biology, prior to the clinical diagnosis of HCM. While our study has highlighted the potential role of ECG changes as indicators for identifying mutation carriers among relatives of individuals with HCM, it is important to recognize the limitations of our findings. The prevalence of ECG changes in the G+/LVH− group was found to be less than 50%, and we acknowledge that a test with low sensitivity may not be ideal as a primary screening tool. The cross-sectional design and relatively small sample size of this study may have contributed to the observed low prevalence of ECG changes, as mutation carriers may not have yet developed these ECG abnormalities. Nevertheless, our findings suggest the possibility of implementing a mutation carrier detection model within families affected by HCM, where ECG could play a central role when combined with other relevant clinical factors. This alternative approach is particularly relevant in developing countries and resource-constrained healthcare settings where routine genetic testing may not be feasible or appropriate. Longitudinal studies on a cohort of G+/LVH− patients are required to demonstrate the variety of phenotypes associated with sarcomere mutations, to understand the relation between early manifestations and disease expression, and to determine how early phenotypes might relate to major consequences of HCM, including arrhythmias and heart failure. Furthermore, such studies are essential for determining the true diagnostic efficacy of ECG changes and for exploring additional complementary screening strategies.
Footnotes
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Phong Dinh Phan. The first draft of the manuscript was written by Phong Dinh Phan, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. Conceptualization: Phong Dinh Phan and Hung Manh Pham; Methodology: Phong Dinh Phan; Formal analysis and investigation: Phong Dinh Phan; Writing - original draft preparation: Phong Dinh Phan; Writing - review and editing: Phong Dinh Phan, Viet Tuan Tran, Minh Nhat Pham, Anh Trung Mai, Dat Tuan An, and Hung Manh Pham; Resources: Hung Manh Pham; Supervision: Hung Manh Pham.
Data availability statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.