
Abstract
Background
The platelet fibrinogen receptor represents the final common pathway of platelet activation, and is formed from two glycoprotein (GP) subunits (GPIIb/IIIa). Carriage of the mutant PlA2 allele of GPIIIa has been shown to confer an increased risk of cardiovascular events, but published studies have disagreed as to the mechanism for this association.
Objectives
To assess whether carriage of the PlA2 allele conforms to Mendelian patterns of expression and to identify whether carriage of the mutant allele modulates platelet function.
Methods
Expression of the PlA2 allele was assessed in both healthy subjects (n = 25) and patients with known coronary artery disease (n = 90) through the development and validation of a liquid chromatography, tandem mass spectrometry (LC-MS/MS) assay. Platelet function was assessed in the patient cohort in response to multiple agonists, and these data were analysed in the context of the proteomic data.
Results
Expression of the wild-type PlA1 allele and mutant PlA2 alleles was readily quantifiable and conformed to Mendelian patterns in both healthy and patient cohorts. Patients who were homozygous for the mutant PlA2 allele had an increased aggregatory response to adenosine diphosphate, collagen, adrenaline, ristocetin, thrombin receptor-activating peptide 6 and U46619, when assessed using agonist-concentration response curves.
Conclusions
These findings support the hypothesis that carriage of the mutant PlA2 allele mediates an increased risk of cardiovascular events through the modulation of platelet reactivity.
Introduction
The fibrinogen receptor is the most abundant integrin on the platelet surface, and represents the final common pathway of platelet activation, adhesion and aggregation.1,2 Formed from two glycoprotein (GP) subunits (GPIIb/IIIa), the receptor binds a combination of fibrinogen, von Willebrand factor (vWF) and fibronectin in a process that terminates haemorrhage following vascular injury.3–5 The GPIIIa subunit is polymorphic with single amino acid substitutions resulting in a number of stable allelic variants, the PlA1/A2 diallelic antigen system being one of the more heavily studied due to its association with neonatal alloimmunity and an increased risk of cardiovascular events.6–8 The prevalence of the PlA2 allele varies between ethnic groups, with a frequency of approximately 1% in Oriental populations, rising to 15% in Caucasian populations.9,10
There had been much disagreement as to the extent, and indeed validity, of any association between carriage of the mutant PlA2 allele and cardiovascular disease until recent meta-analyses showed that carriage of the PlA2 allele does indeed confer a moderate increased risk of both myocardial infarction (n = 40,692; OR 1.08, 95% CI 1.02–1.13; p = 0.004) and ischaemic stroke (n = 11,873; OR 1.12, 95% CI 1.03–1.22; p = 0.011).11,12 Significant heterogeneity was observed across these analyses, and it is unclear whether this represents the challenge of identifying the contribution of a single polymorphism to a multifactorial, polygenic pathological process 13 or variation in an individual’s expression of the PlA1/A2 proteins in heterozygotes. It is not yet known whether expression of the PlA1/A2 proteins obey Mendelian rules or whether subjects identified as genetically heterozygous may in fact have non-uniform patterns of protein expression. A non-uniform pattern of expression would be represented by a heterozygous individual expressing the PlA1/A2 proteins in a ratio that diverges from the 1:1 ratio proscribed by Mendelian rules. If the ratio of expression was found to vary significantly between heterozygous individuals, the utility of genomic techniques to quantify the cardiovascular risk conferred by heterozygous expression would be significantly impaired. If the allele is to realise any potential as a biomarker of cardiovascular disease, either as a standalone test or as part of broader risk stratification, it is vital to understand its proteomic expression.
Uncertainty also remains as to the aetiology of the increased risk conferred by carriage of the PlA2 allele. The mutant allele encodes a single amino acid substitution of proline for leucine adjacent to the ligand binding site, 6 and it has therefore been hypothesised that the polymorphism leads to increased platelet reactivity. Studies in static systems have shown no impact of the polymorphism on ligand binding,14,15 although those performed in cell culture under conditions of shear stress have noted enhanced binding to both fibrinogen and vWF.16,17 The response to platelet agonists has similarly found to be mixed.14,18 It has also been suggested that carriage of the PlA2 allele leads to aspirin resistance, a phenomenon that confers increased cardiovascular risk, 19 although a recent meta-analysis does not support this hypothesis. 20
In this study, we have developed a liquid chromatography, tandem mass spectrometry (LC-MS/MS) assay to measure the expression of platelet PlA1/A2 peptides in both healthy subjects and patients with cardiovascular disease. The use of a LC-MS/MS assay containing internal reference peptides provides a truly quantitative measure of peptide expression. We hypothesise that the proteomic expression of the PlA1/A2 alleles can be quantified using LC-MS/MS, conforms to Mendelian rules of expression, and that increased cardiovascular risk conferred by carriage of the PlA2 allele is secondary to increased levels of platelet reactivity.
Methods
Subject recruitment
Twenty-five clinically healthy subjects, without personal or family history of a bleeding disorder, and having not consumed any medication for at least 14 days, were recruited as the ‘healthy cohort’ to facilitate the development of the LC-MS/MS assay.
Patients with stable coronary artery disease were recruited from Guy’s and St Thomas’ NHS Foundation Trust following an automated search of the TOMCAT clinical database (Phillips) covering the period 6 January 2012 to 11 January 2013. A summary of the recruitment protocol can be seen in Figure 1. Ninety patients were recruited as the ‘patient cohort’, and satisfied the following inclusion criteria: (i) presence of angiographic coronary artery disease and (ii) prescribed daily 75 mg aspirin therapy as their sole antiplatelet agent. Patients were excluded if they had a clinical history suggestive of unstable angina or acute coronary syndrome (ACS) in the previous four months.21,22 Ethical approval for the study was obtained from London-Bloomsbury Research Ethics Committee (Reference: 11/LO/1371). All participants were recruited from King’s College London and Guy’s and St Thomas’ NHS Trust, provided written informed consent to participate in this study, with the consent procedure approved by the ethics committee.
Summary of patient recruitment.
Platelet function testing
Blood was drawn by venipuncture into trisodium citrate (final concentration 0.32%) from an antecubital vein, with samples sent for biochemical and haematological analyses. Whole blood was centrifuged (15 min, 200 × g) at room temperature (RT) to obtain platelet-rich plasma (PRP), with platelet-poor plasma (PPP) obtained by further centrifugation of PRP (5 min, 7000 × g, RT). The platelet count of PRP was not adjusted, and this approach is consistent with current recommendations. 23
Light transmission aggregometry (LTA) was performed in response to arachidonic acid (1.6 mM; AA, Sigma) and adenosine diphosphate (10 µM; ADP, LabMedics) using a PAP8E aggregometer (Bio/Data Corporation) as previously described. 24 Briefly, 225 µL PRP was incubated for 2 min at 37℃, before the addition of 25 µL agonist. Agonist and PRP were continually mixed by a siliconised stirrer bar generating a low-shear vortex (1200 r/min), with measurements of light transmission recorded over 5 min. Light transmission through unstimulated PRP and PPP was calibrated to 0% and 100% aggregation, respectively. Aspirin resistance status in the patient cohort was assigned based on ≥20% aggregation on LTA in response to 1.6 mM AA and/or ≥70% aggregation in response to 10 µM ADP.25,26
Categorisation of platelet function was performed using the Optimul assay, which utilises a similar principle to LTA, but enables multiple agonist concentrations to be assessed simultaneously. 27 Briefly, a 96-well flat-bottomed plate was prepared with platelet agonists to achieve the following concentrations on the addition of 100 µL PRP: AA (0.03–1.6 mM), ADP (0.3–30 µM), collagen (0.1–30 µg/mL; LabMedics), adrenaline (0.001–100 µM; LabMedics), ristocetin (0.18–2 mg/mL; Helena Laboratories), thrombin receptor-activating peptide 6 (0.1–30 µM; TRAP-6, Bachem) and U46619 (0.1–30 µM; Cayman Chemical). On addition of PRP, the plate was immediately placed in a 96-well plate reader (Tecan Sunrise) and absorbance determined at 595 nm every 15 s for 16 min, between vigorous linear shaking at 37℃.
Mass spectrometry
An expanded description on the development and validation of the LC-MS/MS assay is available in the Supplemental Material.
A selective reaction monitoring (SRM) assay was developed to enable the absolute quantification of PlA1/A2 peptides through the use of synthetic peptides as internal reference standards.28,29 The assay was validated in accordance with standard criteria, 30 and the lower limit of measuring range (LMR) observed to be 20 fmol for both wild-type and mutant peptides. Peptide quantification ≥20 fmol could therefore be regarded as an accurate representation of the amount of peptide within the sample.
Briefly, 10 µg platelet lysates from each subject underwent in-gel enzymic digestion before PlA1/A2 peptides were quantified with reference to a 200 fmol spike of isotopically distinct synthetic peptides. 29 Analyses were performed sequentially on a Vantage triple stage quadrupole mass spectrometer (Thermo), with regular quality control samples. Data processing was performed using Pinpoint v1.0 (Thermo), and underwent manual interrogation to ensure the presence of appropriate time-aligned transitions of peptide fragmentation.
Statistical methods
All data are expressed as mean ± standard error (SEM), unless otherwise stated. Analyses of unpaired data were performed using Mann-Whitney test, with analyses of categorical data performed using Fisher’s exact test. The Bonferroni method was used to correct for multiple comparisons. Agonist-concentration response curves were plotted and analysed according to a four parameter logistic equation, where the lower and upper boundaries were set at 0 and 100%, respectively. Area under the curve (AUC) values were calculated using the trapezoid rule from the log-concentration response curve. Analyses of pooled data based on genotype were performed using a Friedman test for paired, non-parametric data. Inter-assay agreement of aspirin resistance status used Cohen’s Kappa (κ). 31 All statistical analyses were performed using GraphPad version 4 (Prism), and significance was taken as p < 0.05 (two-tailed).
Results
The proteomics of platelet PlA1/A2 expression
Expression of the PlA1/PlA2 alleles was inferred through the presence of the corresponding prototypic peptides (PlA1 peptide: DEALPLGSPRC; PLA2 peptide: DEALPPGSPRC). The assignment of genotype based on peptide detection above the LMR enabled healthy subjects to be clearly divided into three groups: expression of only the wild-type peptide (PlA1/A1; n = 16), expression of only the mutant peptide (PlA2/A2; n = 1) and expression of both wild-type and mutant peptides (PlA1/A2; n = 8) (Figure 2). There was no significant difference in the measured expression of PlA1/A2 peptides in heterozygous subjects within the healthy cohort.
Peptide expression in healthy subjects based on inferred genotype.
In the patient cohort, genotyping of subjects based on peptide expression was similarly unequivocal, with 69 patients identified as homozygous for wild-type, two patients homozygous for mutant and 19 patients heterozygous (Table 1). The overall prevalence for carriage of the PlA2 allele was 28% (32% in Caucasian individuals) when the cohorts were combined.
Frequency of PlA2 allele expression.

Biochemical and haematological profiles based on carriage of PlA2 allele.

Significant p values following Bonferroni correction for multiple comparisons.
HbA1c: glycated haemoglobin; HDL: high density lipoprotein; LDL: low density lipoprotein.
Patient characteristics based on carriage of PlA2 allele.

Significant p values following Bonferroni correction for multiple comparisons.
In heterozygous subjects, no significant differences were observed between expression of the PlA1 and PlA2 peptides in healthy subjects (ratio PlA1/PlA2 1.12 ± 0.01; p = 0.245), but a significant difference was observed in the patients cohort (ratio PlA1/PlA2 1.17 ± 0.02, p = 0.023), the ratio of PlA1/A2 expression in heterozygotes being significantly greater in the patient cohort than in the healthy cohort (p = 0.013) (Figure 3).
Box and whisker plot for PlA1:PlA2 peptide expression in heterozygote subjects.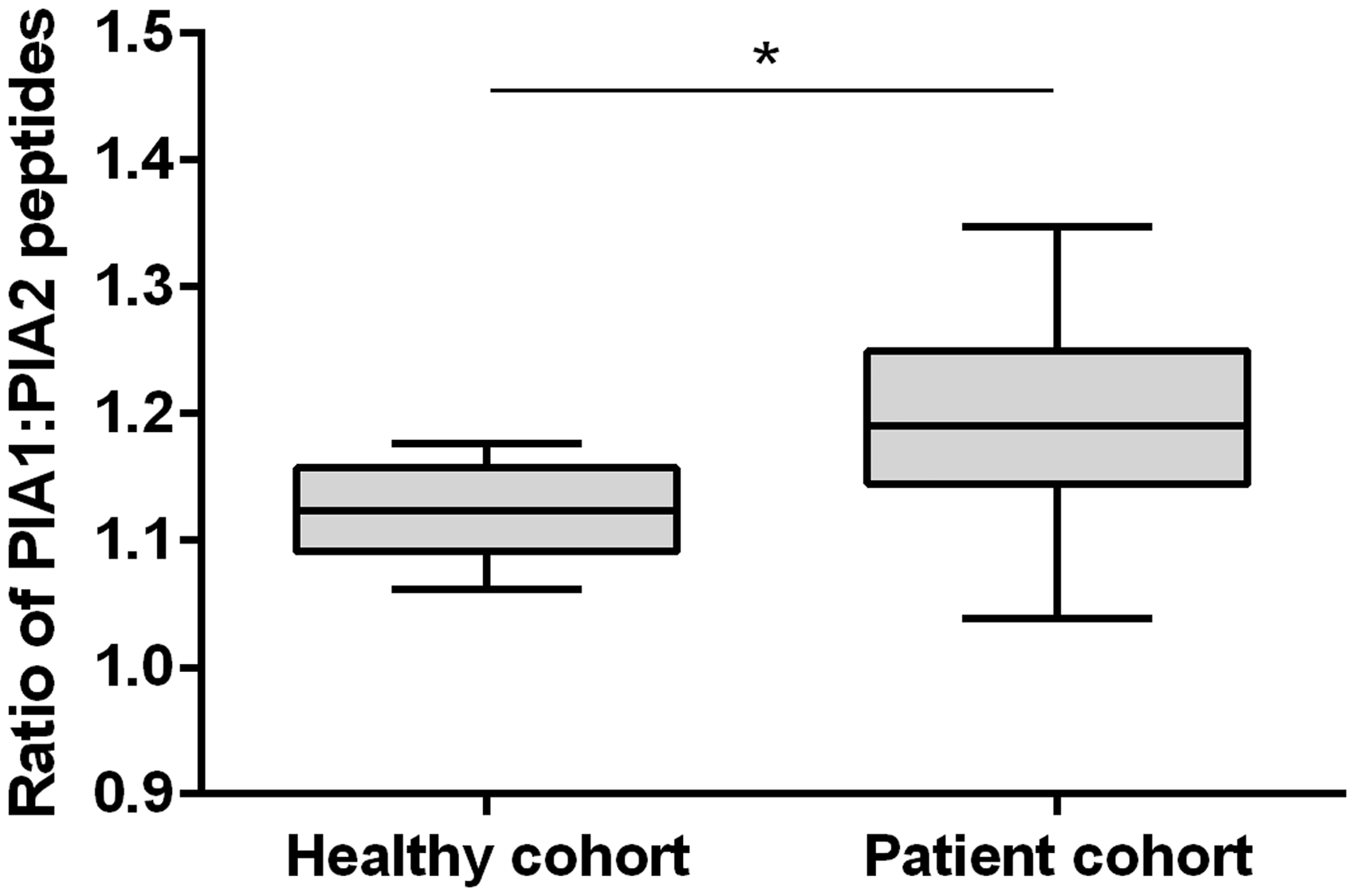
Carriage of the PlA2 allele and platelet function testing in the patient cohort
Analysis of platelet function by Optimul revealed significant differences in aggregation between PlA1 homozygotes and PlA2 homozygotes at higher concentrations of ADP, adrenaline, collagen, ristocetin, TRAP-6 and U46619. Optimul agonist-concentration response curves were plotted according to a four parameter logistic equation for all agonists except AA, where inhibition of platelet aggregation by aspirin therapy did generate sufficient data (Figure 4).
Impact of carriage of the PlA2 allele on platelet function as assessed by Optimul.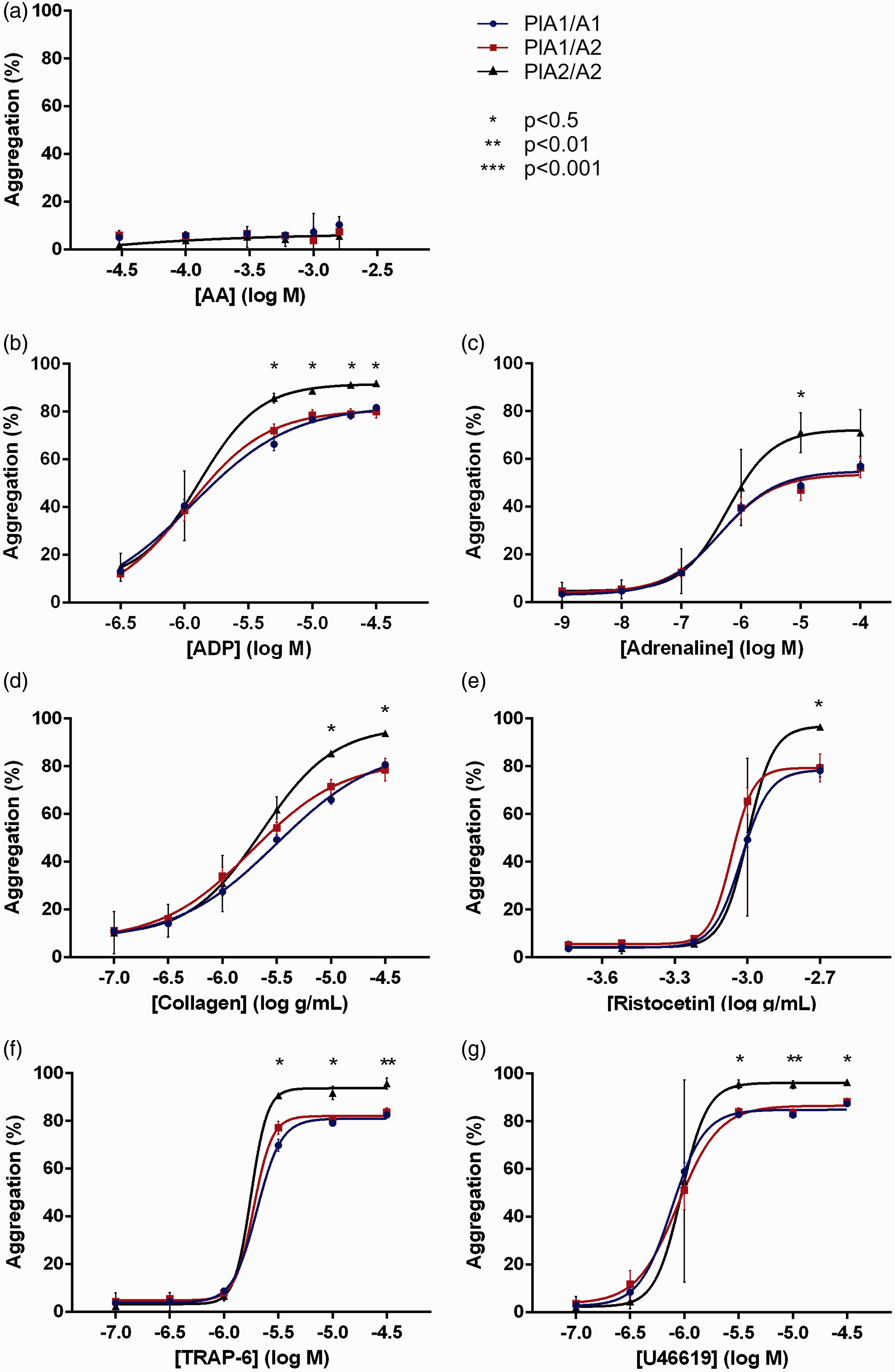
Analysis of agonist-concentration response curves.
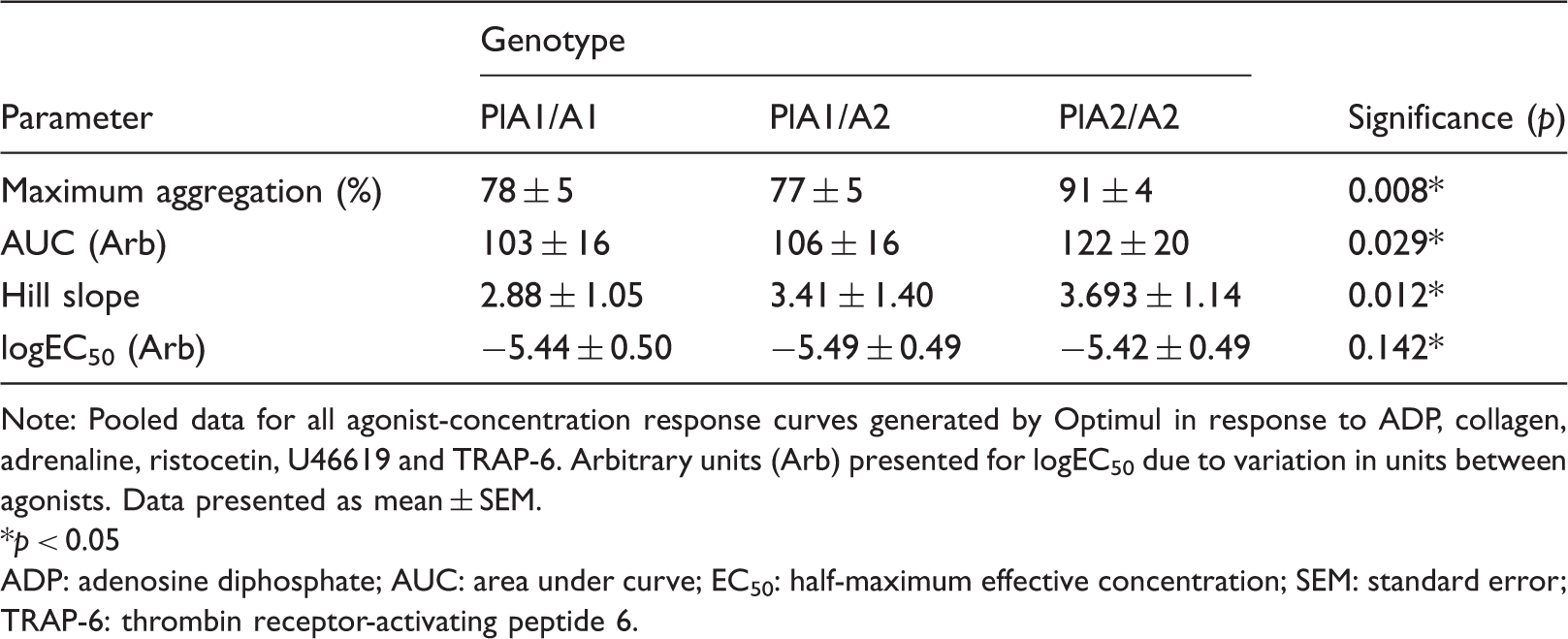
Note: Pooled data for all agonist-concentration response curves generated by Optimul in response to ADP, collagen, adrenaline, ristocetin, U46619 and TRAP-6. Arbitrary units (Arb) presented for logEC50 due to variation in units between agonists. Data presented as mean ± SEM. *p < 0.05
ADP: adenosine diphosphate; AUC: area under curve; EC50: half-maximum effective concentration; SEM: standard error; TRAP-6: thrombin receptor-activating peptide 6.
Association between carriage of the PlA2 allele and aspirin resistance
Twelve patients were identified as aspirin-resistant by AA-induced LTA (13.3%), of which none were carriers of the PlA2 allele. Fourteen patients were identified as aspirin-resistant by ADP-induced LTA (15.6%), of which two were carriers of the PlA2 allele. Inter-assay agreement on the assignment of aspirin resistance status as identified by ADP-induced LTA was poor (κ = 0.192, 95% CI − 0.064 to 0.447).
Discussion
Meta-analyses have shown that carriage of the PlA2 allele of GPIIIa is a risk factor for myocardial infarction and ischaemic stroke, but there remains uncertainty as to the aetiology of this increased risk.11,12 To date, all published studies have assessed PlA1/A2 status using genetic techniques, with the underlying assumption that subjects labelled as heterozygous had equal expression of the corresponding proteins within their platelets in accordance with Mendelian rules of inheritance. However, with the identification of X-chromosome inactivation in females, imprinting of some autosomal genes and preferential expression of certain single nucleotide polymorphisms, it is clear that Mendelian gene expression is not absolute and that carriage of heterozygous alleles does not always result in equal expression of the proteins they encode.32–34 Here, we have developed an LC-MS/MS assay to enable the absolute quantification of platelet PlA1/A2 peptides and to assess whether Mendelian rules of expression do indeed apply to this important polymorphism.
Exploration of the performance characteristics of the assay demonstrated that it was ‘fit for purpose’ to accurately quantify the PlA1/A2 peptides, as measures of both reproducibility and precision conformed to accepted standards. 30 The LMR for both peptides was 20 fmol and, as shown in Figure 2, this facilitated a clear division of genotypes based on the presence of the corresponding peptides. In the healthy cohort, there was no significant difference in the expression of PlA1/A2 peptides in heterozygous subjects clearly indicating that expression does indeed obey Mendelian rules within this cohort.
The data for the patient cohort were less clear in regard to obeying Mendelian expression patterns, as there was a reduced expression of the mutant peptide when compared to the wild-type in heterozygous individuals. It has previously been observed that both cardiovascular disease and aspirin therapy can modulate gene expression. It is therefore possible that either, or both, of these mechanisms may be contributing to a down regulation of the mutant allele and a consequent reduction in cardiovascular risk.35–37 Alternatively, in patients with stable cardiovascular disease their underlying inflammatory phenotype may lead to a preferential activation and subsequent degradation of mutant fibrinogen receptors in heterozygous subjects. 38 However, the wider interquartile range observed in the patient cohort combined with a relatively small difference in the PlA1/PlA2 ratio suggests that these data are more likely to represent variation in sample storage, preparation and analysis between the cohorts. Further investigation in a larger cohort is required to explore the PlA1/PlA2 ratio further.
The prevalence of the PlA2 allele has been previously suggested to be approximately 0.15 in Caucasian populations, based on a study of 200 random Dutch blood donors. 10 Here, we have observed a similar prevalence of the mutant allele in Caucasian patients (0.13), with none of the four Asian patients identified as carriers.
Carriage of the PlA2 allele in the patient cohort gave rise to a demonstrable difference in platelet function as assessed by the Optimul assay, which enabled us to produce a comprehensive assessment of how carriage of the PlA2 allele impacts on platelet function in response to a broad range of agonists. The increase in platelet aggregation for PlA2 homozygotes was shown to be not restricted to a single pathway of platelet activation, but rather was observed all agonists under investigation (other than AA). The demonstrable aspirin-medicated inhibition of platelet COX-1 in all carriers of the PlA2 allele impaired the analysis of how the PlA1/A2 polymorphism may modulate AA-induced aggregation.
The use of Optimul also enabled the plotting of agonist-concentration response curves and the analysis of aggregation responses in their totality rather than solely a comparison of isolated data points. Variation within the patient cohort (including demographics, biochemical/haematological parameters, prescribed medication and co-morbidities) combined with only two PlA2 homozygous subjects meant that analyses of curve parameters were underpowered when considered for each agonist separately. However, the pooling of data for all agonists revealed a significant increase in maximal aggregation, AUC and Hill slope for PlA2 homozygotes.
These findings support the hypothesis that carriage of the PlA2 allele does indeed modulate platelet reactivity through a final common pathway, and the putative mechanism of enhanced ligand binding in the presence of this mutation is compatible with the observed increase in Hill slope. The significant increase in aggregation was only observed for PlA2 homozygotes, but we hypothesise that a moderate increase in aggregation would also be observed for heterozygotes within an appropriately powered data set. The difference in aggregation response cannot be explained by either differing haematological/biological profiles or patient characteristics. However, the identification of only two PlA2 homozygotes is a clear limitation of this study, and a larger dataset would be required to corroborate these findings.
Carriage of the PlA2 allele was not observed to confer an increased risk of aspirin resistance, defined by either AA-induced or ADP-induced LTA. This is perhaps unsurprising given the importance of shear forces to the phenotype determined by the polymorphism and indicates substantial issues with most previously published studies investigating the association between aspirin resistance and this polymorphism. 20 The poor inter-assay agreement on aspirin resistance status further highlights the controversies surrounding the precise identification of this phenomenon. 39
Aside from the findings presented here, this work raises a broader issue about how one should approach the analysis of platelet function within a clinical environment. LTA is the current ‘gold standard’, but in previously published work appears to provide an inaccurate assessment of platelet function for carriers of a common genetic polymorphism. These findings may be the result of differences in assay mechanics when compared to Optimul or possibly represent the limitations of a technique that precludes plotting agonist-concentration response curves for multiple agonists. Further investigation in a healthy population should further categorise of how the choice of platelet function assay may impact on the observed effect of the polymorphism.
Conclusions
Platelet PlA1/A2 peptides are readily quantifiable by LC-MS/MS and appear to obey the rules of Mendelian expression, with homozygosity for the mutant allele appearing to confer a global increase in platelet reactivity. The pairing of proteomic and platelet function data as we have demonstrated here has potential for the future understanding of platelet aggregation responses.
Footnotes
Acknowledgements
The authors would like to acknowledge the technical assistance for the mass spectrometry provided by Proteome Sciences plc.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The work presented here was funded by Guy’s and St Thomas’ Charity (Registered Charity no. 251983).
Ethical approval
Ethical approval for the study was obtained from London-Bloomsbury Research Ethics Committee (Reference: 11/LO/1371).
Guarantor
AF is the guarantor for all the content presented in this paper.
Contributorship
All authors conceived the study question, and CNF was responsible for the data collection. All authors were involved in the design phase, the analysis and the interpretation of the results. CNF drafted the manuscript, and all authors approved the final version.