
Abstract
We report a rare case of acquired, transient platelet dysfunction induced by infection in an older patient. An 84-year-old female with persistent pneumonia presented with severe mucocutaneous bleeding and anemia (nadir hemoglobin: 38 g/L) despite normal platelet count and coagulation parameters. Platelet function tests showed absent or reduced aggregation to all standard agonists. Treatment included platelet transfusion support, targeted anti-infection therapy, and adjunctive agents (ulinastatin and sulodexide) to address inflammation and endothelial dysfunction. The patient’s platelet function normalized with the resolution of infection. This case highlights infection as an underrecognized etiology of acquired platelet dysfunction, particularly in older patients, where normal platelet counts may obscure the diagnosis of major platelet functional impairment.
Introduction
Acquired platelet dysfunction (APD), manifesting as unexplained bruising or mucosal bleeding, is most frequently caused by medications that directly interfere with platelet function. 1 However, the epidemiology and mechanisms by which complex systemic disorders induce platelet dysfunction are poorly characterized. Emerging evidence indicates that platelets have diverse functions beyond hemostasis, including inflammation modulation.2,3 Experimental studies in community-acquired pneumonia have validated platelet activation by specific antigen, and this inflammatory engagement during infection may lead to platelet dysfunction. 4 Yet, most clinical studies have focused on changes in circulating platelet counts rather than on functional impairment. In sepsis and critical illness, the prevalence of thrombocytopenia ranges from 20% to 50%. Functional platelet defects have been reported to correlate with sepsis severity, but their frequency and clinical significance in older populations remain unclear. 3
Diagnosis and management of APD are challenging due to heterogeneity of clinical presentations, ambiguity in etiology and pathogenesis, technical limitations in platelet function tests, and lack of well-established protocols.1,5 Here, we report a case of acquired, transient platelet dysfunction induced by infection that resulted in severe anemia and hemorrhage. We also describe the novel, empirical use of ulinastatin and sulodexide as adjunctive agents targeting inflammation and endothelial dysfunction.
Case report
An 84-year-old, bedridden female with a history of cervical spinal cord injury and high-level paraplegia following traumatic injury 10 months earlier was referred to our hospital for severe anemia and mucocutaneous bleeding. One month prior to the current admission, she had been hospitalized at another facility for aspiration pneumonia. Sputum culture grew Pseudomonas aeruginosa; treatment initially included netilmicin, and meropenem was subsequently added because of persistent fever. Her infection was partially contained (body temperature: 37 ℃–38 ℃), while she developed progressive anemia (nadir hemoglobin: 52 g/L) with new-onset purpura, gingival bleeding, and melena.
The patient denied personal or familial bleeding history. Records from her previous cervical spinal cord surgery revealed no significant perioperative hemorrhage. Her medical history was notable for coronary artery disease, paroxysmal atrial fibrillation, and chronic heart failure; she had been receiving edoxaban (30 mg qd, discontinued upon the onset of bleeding). She also had hypertension treated with sacubitril/valsartan (100 mg qd), and type 2 diabetes mellitus treated with insulin lispro (6 U before meals) and insulin glargine (4 U at bedtime). On examination, the patient was alert and oriented. The temperature was 37 ℃, pulse 75 beats/min, respiratory rate 18 breaths/min, and blood pressure 126/60 mm Hg. She had gingival bleeding and scattered purpura on her lower limbs.
On admission, blood investigations showed severe normocytic normochromic anemia (hemoglobin: 41 g/L, mean corpuscular volume: 91.6 fL) with normal platelet count (221 × 109/L). Hemostatic screening tests were normal (prothrombin time: 12.3 s, activated partial thromboplastin time: 24.3 s, thrombin time: 17.9 s, for control values of 13.4, 38.1, and 17.8 s, respectively; Table 1). Bone marrow cytomorphology abnormalities or von Willebrand factor (VWF) deficiency was not detected. Renal and liver function were also normal. Gastroscopy and colonoscopy found no active hemorrhage, suggesting a small-bowel origin of gastrointestinal bleeding. Enteroscopy was refused by the patient and her family because of potential risks. Despite 2 weeks of anti-anemic therapy (iron sucrose, folic acid, mecobalamin, and transfusion of 7 units of packed red blood cells), together with initiation of hemostatic medications including carbazochrome sodium sulfonate (80 mg qd), tranexamic acid (0.5 g bid), and etamsylate (2.5 g bid), her hemoglobin declined to 38 g/L (Figure 1).
Serial laboratory test results.
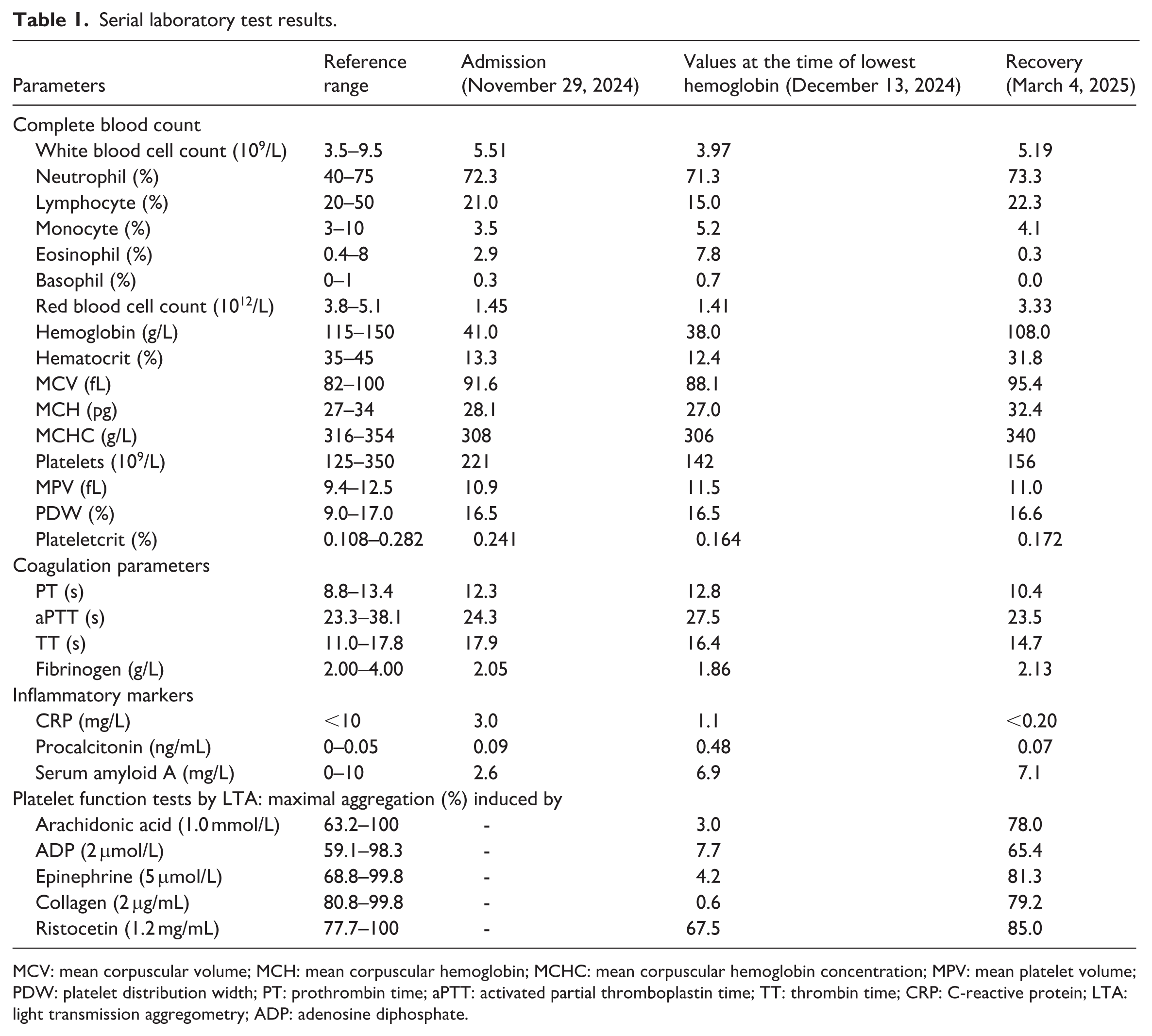
MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin; MCHC: mean corpuscular hemoglobin concentration; MPV: mean platelet volume; PDW: platelet distribution width; PT: prothrombin time; aPTT: activated partial thromboplastin time; TT: thrombin time; CRP: C-reactive protein; LTA: light transmission aggregometry; ADP: adenosine diphosphate.
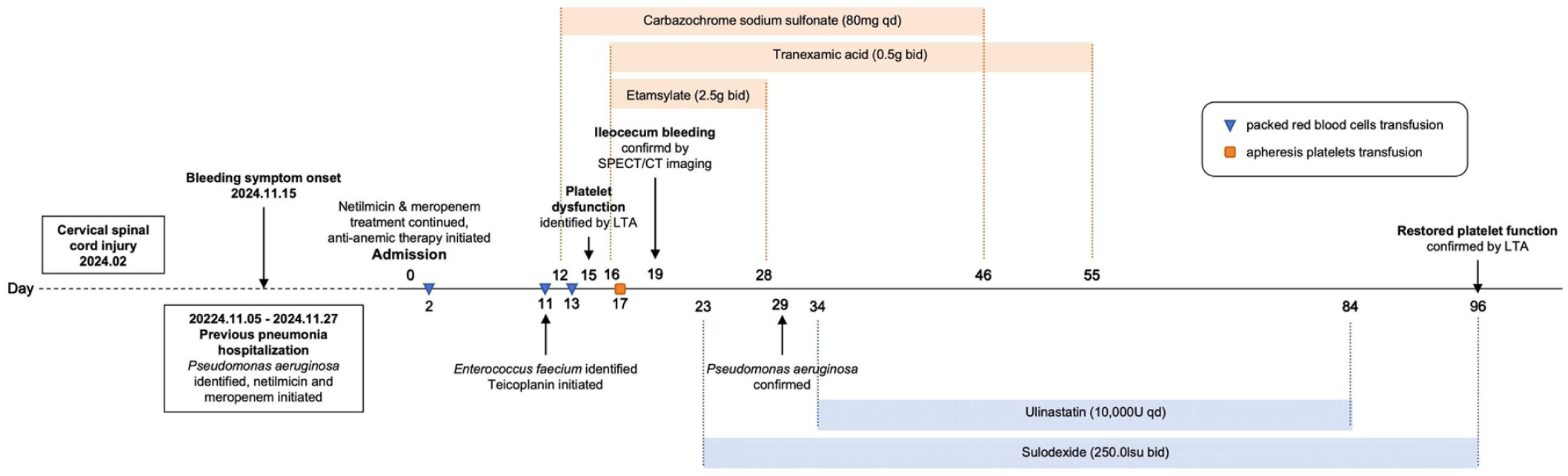
Timeline of major clinical events and interventions.
Platelet function testing by light transmission aggregometry (LTA; December 13, 2024) revealed severe platelet dysfunction. A panel of five agonists (arachidonic acid: 1.0 mmol/L, adenosine diphosphate (ADP): 2 μmol/L, epinephrine: 5 μmol/L, collagen: 2 μg/mL, ristocetin: 1.2 mg/mL) was used. Aggregation was absent with arachidonic acid (max: 3.0%, normal range: 63.2%–100%), collagen (max: 0.6%, normal range: 80.8%–99.8%), and epinephrine (max: 4.2%, normal range: 68.8%–99.8%). Aggregation was markedly reduced with ADP (max: 7.7%, normal range: 59.1%–98.3%). Aggregation curves showed an initial shape change followed by no aggregation response to ADP (Figure 2). There was only decreased aggregation to ristocetin (max: 67.5%, normal range: 77.7%–100%; Figure 3). Repeat LTA at an independent laboratory confirmed these findings. Flow cytometry showed no abnormalities in GPIIb (99.97%), GPIIIa (99.95%), or GPIb (100.00%; GPIIb, GPIIIa, GPIb normal range: 90.00%–100.00%). To exclude hereditary etiologies, targeted next-generation sequencing using a comprehensive hematologic-disease gene panel covering 1080 genes was performed. No pathogenic or likely pathogenic germline variants associated with hereditary platelet disorders were identified, supporting a diagnosis of APD. SPECT/CT (single photon emission computed tomography/ computed tomography) imaging in 99mTc-RBC gastrointestinal bleeding scintigraphy further localized mild but persistent bleeding in the ileocecum.

Platelet aggregation curves on first diagnosis (December 13, 2024) and after treatment (March 4, 2025). Platelet reactivity to arachidonic acid (1.0 mmol/L), ADP (2 μmol/L), epinephrine (5 μmol/L), collagen (2 μg/mL), and ristocetin (1.2 mg/mL) was assessed by light transmission aggregometry. (a) Severe platelet dysfunction on December 13, 2024. (b) Platelet function restored on March 4, 2025.

Maximal platelet aggregation responses by light transmission aggregometry on first diagnosis (December 13, 2024) and after treatment (March 4, 2025). Dashed lines indicate the lower limits of normal platelet aggregation responses.
After establishing the diagnosis of severe anemia and ileocecum hemorrhage caused by APD, we initiated platelet function supportive measures along with ongoing hemostatic and anti-anemic therapy. One unit of apheresis platelets was transfused. Ulinastatin (10,000 U qd) and sulodexide (250.0 lsu bid) were prescribed. Netilmicin (0.4 g qd) and meropenem (1 g q8 h) were continued on transfer to our hospital, and a repeat sputum culture (December 27, 2024) grew P. aeruginosa. A urine culture (December 9, 2024) grew Enterococcus faecium, and teicoplanin (0.4 g q12 h for the first three doses, then 0.4 g qd) was added. These antibiotics were given intermittently during the clinical course. With amelioration of infection, the patient’s mucocutaneous bleeding resolved completely. Serial hemoglobin monitoring showed a gradual increase, reaching 108 g/L after 3 months (Table 1). An LTA (March 4, 2025) confirmed improved platelet function. Maximal aggregation to all five agonists was within normal ranges (arachidonic acid: 78.0%, ADP: 65.4%, epinephrine: 81.3%, collagen: 79.2%, ristocetin: 85.0%; Figure 3). Aggregation curves showed slightly reversible aggregation (Figure 2).
Discussion
Reports of acquired, transient platelet dysfunction in older adults are uncommon. APD is potentially underestimated, especially in older adults who have limited hemostatic reserve and are prone to refractory infections, polypharmacy, and multimorbidity. Normal platelet counts and coagulation tests often deter further investigation into the cause of bleeding. This could be attributed to limited access to LTA, which remains the gold standard for platelet function testing. LTA is time-consuming, experience-dependent and poorly standardized, limiting its routine clinical use. 5 Platelet function testing by LTA should be considered when an older patient presents with suspected platelet dysfunction, particularly in the face of a hemostatic challenge like surgery.
As reported in previous cases, APD is commonly induced by medications, hematologic malignancies, and conditions such as APD with eosinophilia, and could present with concomitant thrombocytopenia.6–8 In this case, the varied platelet aggregation responses to different agonists suggested a preserved VWF–GPIb axis but a broad defect in platelet activation, signaling, or secretion. Ristocetin elicits VWF-dependent agglutination via GPIb and is relatively independent of activation or secretion pathways. The preserved response to ristocetin matched normal VWF testing and GPIb expression, and argued against Glanzmann thrombasthenia, which was further excluded by normal GPIIb/IIIa expression and negative genetic testing. By contrast, responses to arachidonic acid, collagen, epinephrine, and ADP probe thromboxane-dependent, GPVI/GPIaIIa-linked, adrenergic and P2 receptor-mediated activation pathways, respectively. Initially reduced aggregation to these agonists, with subsequent restoration upon recovery, was consistent with an acquired, reversible impairment of platelet activation or secretion, potentially associated with broad infection-related inflammatory processes. 5 These findings, together with a thorough medication review, normal coagulation parameters, unremarkable bone marrow examination, and absence of hematologic malignancy, excluded alternative causes, including drug-induced platelet inhibition, inherited platelet disorders, bone marrow disease, and consumptive coagulopathy, supporting infection as the etiology. The temporal association between infection and platelet dysfunction, and the restoration of platelet function with the resolution of infection, further supported this hypothesis.
During infection, an excessive and unbalanced inflammatory response may impair platelet function through multiple mechanisms, including cytokine-mediated platelet activation and exhaustion, endothelial injury, disruption of platelet-endothelium interactions, and immune-mediated platelet destruction. 2 van Gils et al. described the role of platelets in host immune responses to infection, including platelet adhesion that endows the endothelium with a proinflammatory phenotype, thereby amplifying leukocyte recruitment and inflammatory signaling. 9 Sustained inflammatory activation may subsequently lead to platelet hypo-responsiveness and functional impairment.
In light of this pathophysiology, we administered ulinastatin and sulodexide, aiming to attenuate inflammation and protect endothelial function. Ulinastatin, a multifunctional serine protease inhibitor, reduces inflammatory cytokines, neutrophil infiltration, and endothelial dysfunction. 10 A randomized, placebo-controlled pilot study demonstrated that intravenous ulinastatin reduced mortality in severe sepsis. 11 The PANDA trial also reported reduced systemic inflammatory response syndrome rates with ulinastatin in patients undergoing acute aortic dissection surgery, suggesting a protective effect against deleterious inflammatory response. 12
Sulodexide, a heparin-like glycosaminoglycan, exerts anti-inflammatory effects by inhibiting leukocyte activation and downregulating the secretion of inflammatory mediators. 13 It helps preserve and restore the endothelial glycocalyx, which is critical for vascular permeability, shear stress responses, and platelet–endothelium interactions. A pilot study in type 2 diabetes mellitus found that sulodexide restored glycocalyx perturbation and vascular permeability. 14 A randomized, placebo-controlled trial in convalescent COVID-19 patients reported reduced serum endothelial dysfunction biomarker levels with sulodexide. 15
Conclusions
The concurrent resolution of infection and normalization of platelet function suggests infection-induced platelet dysfunction, though the relative contributions of antimicrobial therapy, platelet transfusion, and adjunctive agents cannot be differentiated in this single case. Future studies are warranted to clarify the mechanisms of infection-induced platelet dysfunction and to evaluate the therapeutic potential of anti-inflammatory and endothelial-protective strategies (Supplemental Material).
Supplemental Material
sj-pdf-1-sco-10.1177_2050313X261436132 – Supplemental material for Acquired platelet dysfunction induced by infection in an older patient: Severe hemorrhage despite normal platelet count
Supplemental material, sj-pdf-1-sco-10.1177_2050313X261436132 for Acquired platelet dysfunction induced by infection in an older patient: Severe hemorrhage despite normal platelet count by Yuanyuan Jiang, Nan Lu, Xinmiao Chang, Yanlan Guo, Jinlong Liu, Yinlian Ru and Wenbin Wu in SAGE Open Medical Case Reports
Footnotes
Ethical considerations
Ethical approval to report this case was obtained from the Beijing Hospital Ethics Committee (2025BJYYEC-KY154-01). This case report was prepared in accordance with the CARE (CAse REport) guidelines.
Consent to participate
Informed consent was taken from the patient before publishing the case report.
Author contributions
Yuanyuan Jiang: conceptualization; formal analysis; writing – original draft. Nan Lu: conceptualization; formal analysis; writing – original draft. Xinmiao Chang: conceptualization; formal analysis; writing – review and editing. Yanlan Guo: conceptualization; investigation; writing – review and editing. Jinlong Liu: conceptualization; investigation; writing – review and editing. Yinlian Ru: conceptualization; investigation; writing – review and editing. Wenbin Wu: conceptualization; supervision; writing – review and editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by China National Key R&D Program (2020YFC2009006).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.