
Abstract
Background
Endothelial dysfunction is central to the pathogenesis of coronary artery disease, but the role of local and circulating endothelial progenitor cells in maintaining vascular health is poorly understood. We hypothesised that impaired local and circulating vascular repair mechanisms predispose to endothelial dysfunction and the premature onset of coronary artery disease.
Methods and results
Patients with premature coronary artery disease (n = 16) and healthy age- and sex-matched controls (n = 16) underwent venous occlusion plethysmography with intra-arterial infusion of acetylcholine and sodium nitroprusside. Numbers of circulating endothelial progenitor cells were directly quantified in whole blood by flow cytometry. Endothelial cells were isolated from the blood vessel wall and from peripheral blood mononuclear cells, and expanded in vitro for phenotypic and functional characterisation and analysis of microRNA expression levels. A dose-dependent increase in forearm blood flow (p < 0.001) was attenuated in response to the endothelial-dependent vasodilator acetylcholine in patients compared with controls (p = 0.03). No differences in the number of circulating endothelial progenitor cells or in the phenotype, function or microRNA expression levels of endothelial outgrowth cells isolated from blood were observed in patients and controls. Conversely, local vessel wall endothelial cells from patients had significant impairments in proliferation, adhesion and migration, and significantly reduced expression levels of microRNAs known to regulate endothelial function (miRs −10 a, −let7b, −126 and −181 b) (p < 0.05 for all).
Conclusion
Local vessel wall derived endothelial cells, rather than circulating endothelial progenitor cells and their progeny, are impaired in patients with vascular dysfunction and premature coronary artery disease.
Translational perspective
Endothelial progenitor cells (EPCs) hold great therapeutic potential for vascular regeneration, although their origin, whether the bone marrow or blood vessel wall, remains unclear. In order to exploit the full potential of EPCs, it is vital that the cell-specific mechanisms that contribute to the pathophysiology of coronary artery disease are fully understood. Our observation that vessel wall endothelial cells are broadly dysregulated in patients with premature coronary artery disease whereas circulating EPCs are functionally normal, highlights important differences in endothelial cells from different locations. Future studies of vessel wall endothelial cells are likely to advance our understanding of the mechanisms of premature coronary artery disease and may lead to novel approaches to enhance vascular repair.
Introduction
Atherosclerosis occurs as a result of recurrent injury to the vasculature throughout life, with many risk factors known to play a role in the development, progression and clinical consequences of disease.1,2 Our understanding of the cellular mechanisms that underpin coronary artery disease is incomplete. Recent interest in vascular repair and regeneration has gathered momentum, as deficiencies in these mechanisms have been implicated in the pathogenesis of atherosclerosis. 3
Postnatal blood vessel growth and repair was traditionally thought to occur exclusively from the migration and proliferation of endothelial cells within existing mature blood vessels. 4 In 1997, CD34 + EPCs in the adult circulation were reported to promote therapeutic angiogenesis in experimental ischaemia. 5 It is now accepted that early reports of adult EPCs refer to pro-angiogenic cells of haematopoietic origin. 6 As such, a large body of research is dedicated to delineation of the origin, phenotype, function and translational aspects of a true adult human EPC. 7 EPCs are most commonly studied by direct enumeration in peripheral blood by flow cytometry as CD34 + CD133 + KDR + cells. 8 Colony forming assays following ex vivo culture of peripheral blood mononuclear cells (MNCs) are also widely used for studies of EPCs. Late endothelial outgrowth cells (EOCs) lack expression of haematopoietic and leukocyte markers, demonstrate hierarchical proliferative potential and can form de novo blood vessels in vivo.9–12 Therefore, EOCs appear to arise from true circulating EPCs.
Functional heterogeneity has been demonstrated in endothelial cells from different vascular locations, and appears to persist upon removing them from their microenvironment for in vitro analyses.13–16 Therefore it is important to study organ-appropriate endothelial cells, although in humans such studies are limited due to problems with accessibility. Reduced numbers of circulating EPCs have been reported in patients with coronary artery disease.17–24 However, these data are confounded by discrepancies in the phenotypic definitions of EPC with overlap between haematopoietic stem cells and other circulating lineages. 9 Moreover, most studies have failed to confirm that their reported putative EPCs are bona fide progenitor cells, that is, via isolation for in vitro expansion and functional assessment.
We hypothesised that impaired local and circulating vascular repair mechanisms predispose to endothelial dysfunction and the premature onset of coronary artery disease. Here, we have used a broad range of quantitative and functional experiments to assess morphology, growth kinetics, phenotype, function and microRNA expression levels in endothelial cells derived from the vessel wall and from circulating mononuclear cells in patients with premature coronary artery disease and age- and sex-matched healthy control subjects. We demonstrate that significant functional impairments and reduced expression levels of microRNAs known to regulate endothelial cell function are specific to local vessel wall endothelial cells in patients with premature coronary artery disease. Conversely, we observed no differences in the number of circulating EPCs, or in the function and microRNA expression by late outgrowth EOCs isolated from the same patients compared with healthy subjects.
Methods
Detailed description of the materials and methods are available in the Supplementary Material online.
Subjects
The study was performed with the approval of the South East Scotland Research Ethics Committee, in accordance with the Declaration of Helsinki and with the written informed consent of all participants. Patients with premature coronary artery disease and a family history of premature coronary artery disease (n = 16) were identified from the outpatient department, Royal Infirmary of Edinburgh, Scotland, UK. A control group of healthy age- and sex-matched subjects (n = 16) with no evidence of significant coronary artery disease following computed tomography coronary angiography (CTCA) was recruited from the Clinical Research Imaging Centre, Royal Infirmary of Edinburgh.
Tissue sampling and vascular assessment
Subjects attended the Clinical Research Facility at the Royal Infirmary of Edinburgh for vascular assessment and tissue sampling; peripheral blood was collected for direct quantification of endothelial progenitor populations by flow cytometry, and for the isolation and in vitro expansion of circulating EPCs to generate endothelial outgrowth cells. Vessel wall endothelial cells were isolated by wire biopsy for in vitro expansion.
In vitro assessment and microRNA studies
Vessel wall endothelial cells and endothelial outgrowth cells from patients and controls underwent morphological and phenotypic assessment by flow cytometry and immunocytochemistry. Functional assessment of growth kinetics, adhesion and migration, and microRNA extraction and TaqMan qPCR analysis was also carried out in these cells.
Statistical analyses
Plethysmographic data were analysed as described previously. 25 Forearm blood flow (FBF) was analysed using two-way repeated measures analysis of variance. Continuous variables are reported as mean and standard error of the mean (SEM) or median (interquartile range (IQR)). Summary statistics were compared by Chi-squared, Fisher’s exact, Student’s t- and Mann–Whitney U tests where appropriate. Statistical significance was taken at p < 0.05. Statistical analyses were performed with GraphPad Prism (version 6, GraphPad Software Inc., CA, USA).
Results
Characteristics of patients with premature coronary artery disease and controls.
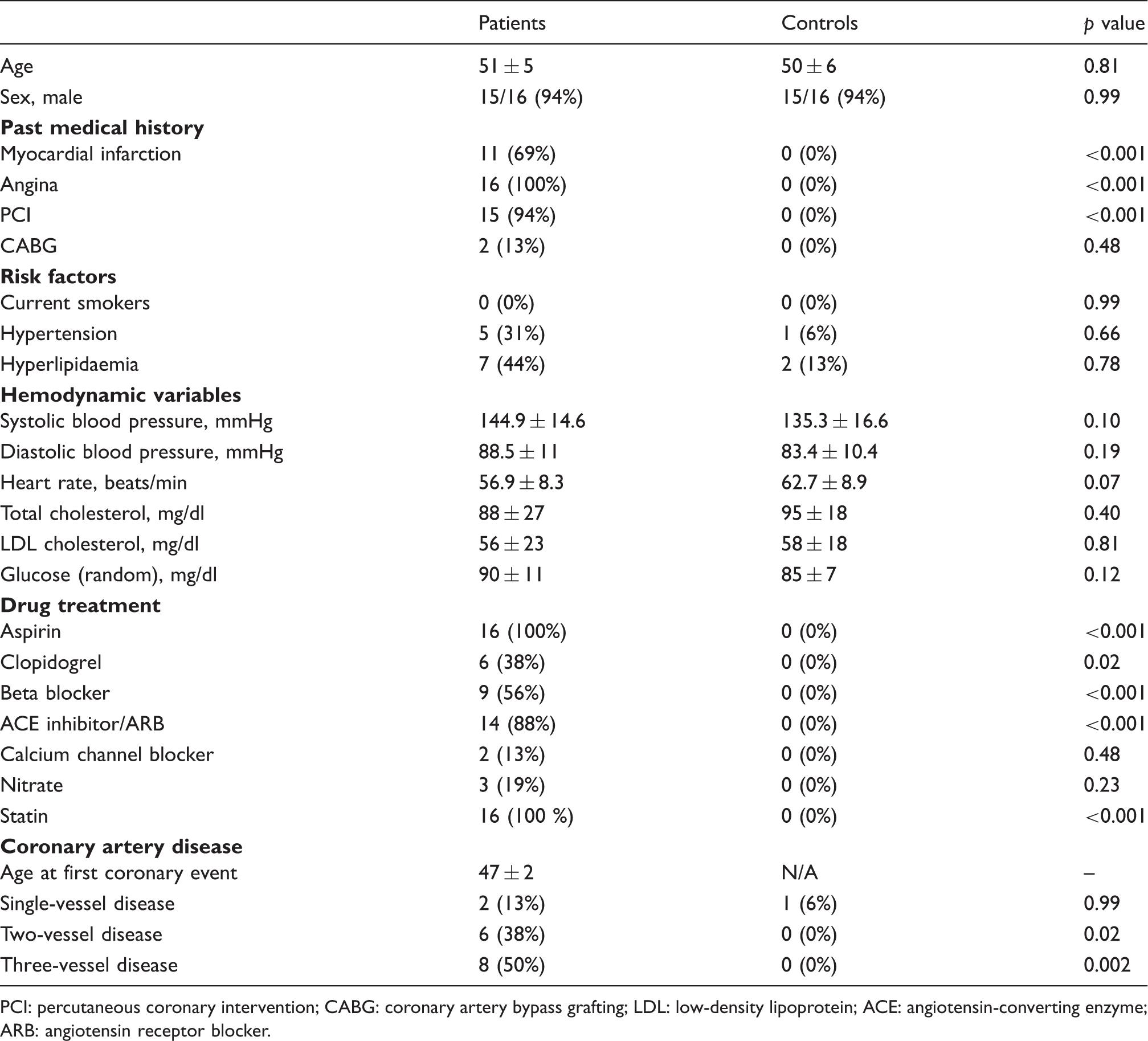
PCI: percutaneous coronary intervention; CABG: coronary artery bypass grafting; LDL: low-density lipoprotein; ACE: angiotensin-converting enzyme; ARB: angiotensin receptor blocker.
Endothelial-dependent and -independent vasodilatation
FBF was measured using venous occlusion plethysmography before and during intra-arterial infusions of endothelial-dependent (acetylcholine) and -independent (sodium nitroprusside) vasodilators. There was a dose-dependent increase in FBF with both vasodilators (p < 0.001). Vasodilatation was reduced in response to acetylcholine in patients compared with controls (peak FBF: 5.1 ± 0.9 vs. 9.3 ± 2.7 ml/100ml of tissue per min, p = 0.03), but was similar in response to sodium nitroprusside (p = 0.37) (Figure 1).
Endothelial-dependent and -independent vasodilatation in patients with premature coronary artery disease and matched controls. Both vasodilators caused a dose-dependent increase in forearm blood flow (FBF) in the infused arm (p < 0.001), but vasodilatation was reduced in response to infusion of the endothelial-dependent vasodilator acetylcholine in patients (red) compared with controls (blue, peak FBF: 5.1 ± 0.9 vs. 9.3 ± 2.7 ml/100ml tissue per min, p = 0.03). Vasodilatation was similar in response to infusion of the endothelial-independent vasodilator sodium nitroprusside (SNP) in patients and controls (p = 0.37, n = 16 for both). Closed circle: infused FBF; open circle: non-infused FBF.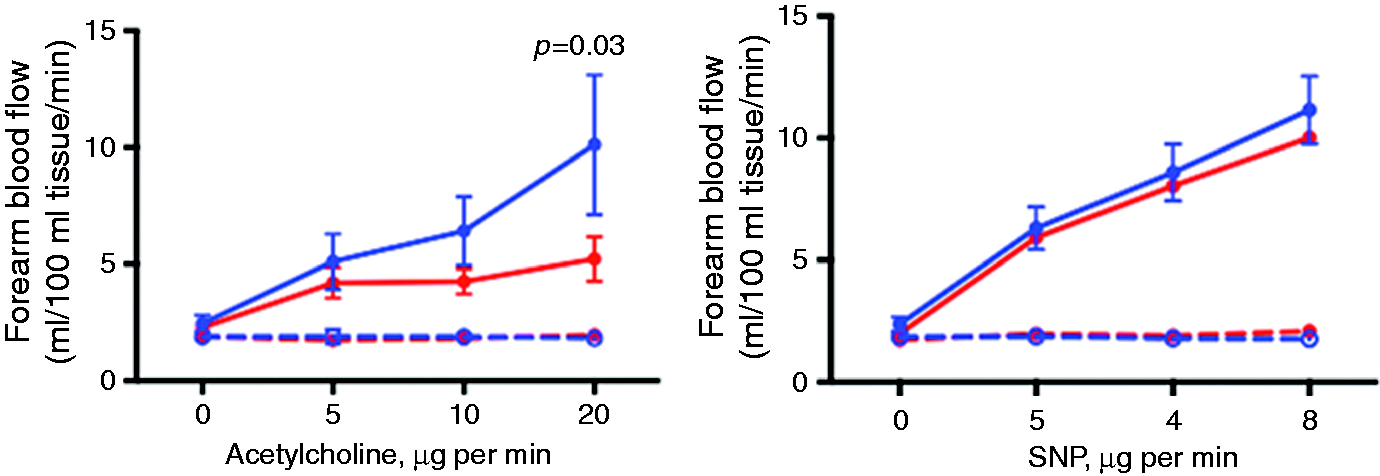
Circulating EPCs
Circulating subpopulations of progenitor cells.
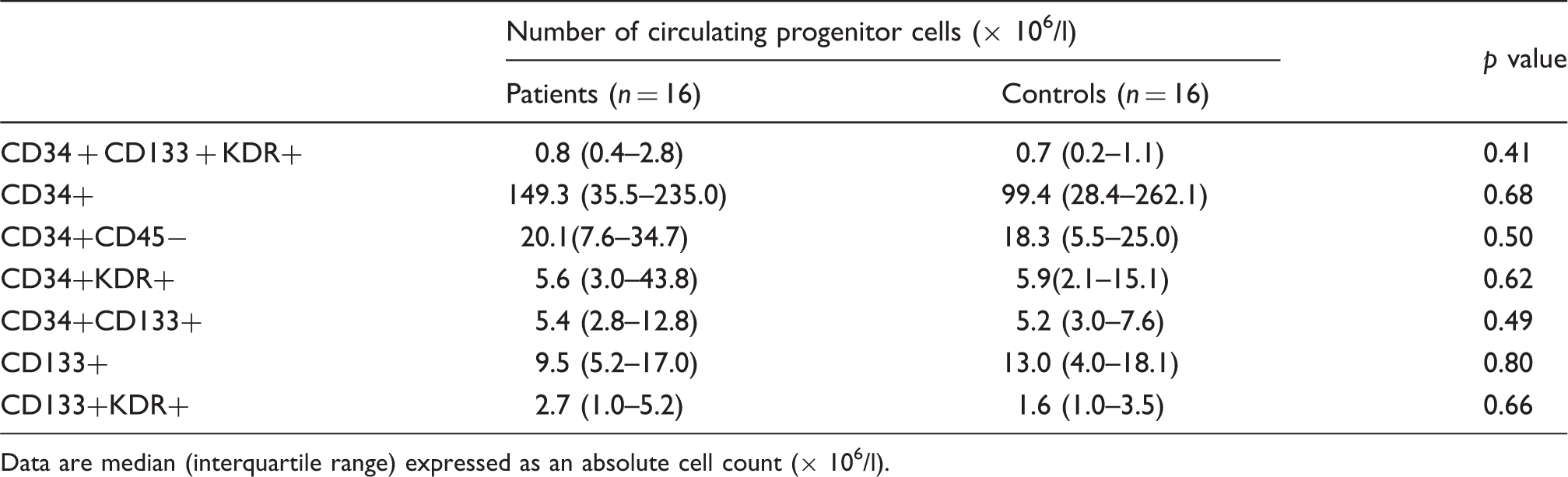
Data are median (interquartile range) expressed as an absolute cell count (× 106/l).
Efficiency of cell isolation and growth kinetics
Growth kinetics of outgrowth from vessel wall endothelial cells and peripheral blood MNCs, that is, late outgrowth EOCs, were calculated to identify functional differences between these cells in patients and controls. Outgrowth was observed as colonies that were expanded to form cobblestone monolayers with typical endothelial morphology, which were maintained in culture up to passage 10 (Figure 2(a) to (c) and (i) to (k)). For vessel wall endothelial cells, colony outgrowth was observed from 14/16 patients (88%) and 15/16 controls (94%), and no differences were observed between patients and controls in the day of first colony appearance (9.1 ± 1.1 vs. 7.4 ± 1.2 days, p = 0.30) or the total number of colonies (10.8 ± 3.3 vs. 9.4 ± 2.5, p = 0.92) (Figure 2(e) and (f)). For late outgrowth EOCs, colonies were derived from all subjects with colonies emerging slightly later from patients compared with controls (10.3 ± 0.8 vs. 7.9 ± 0.5 days, p = 0.03), but with no difference in the number of colonies obtained from patients compared with controls (11.8 ± 2.3 vs. 20.9 ± 17.7 colonies, p = 0.06) (Figure 2(m) and (n)).
Morphology and growth kinetics of vessel wall derived endothelial cells and endothelial outgrowth from circulating cells. Colonies were derived with a high efficiency from vessel wall endothelial cells (ECs) ((a) to (c), × 10 magnification) and from peripheral blood mononuclear cells ((i) to (k), × 10 magnification) in patients and controls. Cell outgrowth from both sources was morphologically analogous with typical endothelial ‘cobblestone’ morphology. Representative images of the emergence and expansion of colonies from a patient with premature coronary artery disease are shown. Vessel wall endothelial cells (d) and endothelial outgrowth cells (EOCs) (l) from patients and controls had ubiquitous expression of von Willebrand factor (vWF; red cytoplasmic staining; DAPI, blue nuclear staining. Images shown are from a control subject at passage 3, scale bars 50 µm). For vessel wall endothelial cells, no differences were observed between patients and controls in the day of first colony appearance (e) (9.1 ± 1.1 vs. 7.4 ± 1.2 days, p = 0.30) or the total number of colonies per subject (f) (10.8 ± 3.3 vs. 9.4 ± 2.5, p = 0.92). For late outgrowth EOCs, colonies emerged later from patients compared with controls (m) (10.3 ± 0.8 vs. 7.9 ± 0.5 days, p = 0.03) but no difference in colony number was observed from patients compared with controls (n) (11.8 ± 2.3 vs. 20.9 ± 17.7 colonies, p = 0.06). Mean population doubling times (PDT) between passages 2 and 8 by vessel wall endothelial cells from patients were significantly reduced compared with controls (g) (mean PDT ± SEM: 2.3 ± 0.2 vs. 3.4 ± 0.5, p = 0.02). Cumulative population doubling levels (CPDL) of vessel wall endothelial cells from patients were significantly reduced compared with controls (h) (CPDL at passage 8: 18.1 ± 1.4 vs. 23.6 ± 1.4, p = 0.03). No significant difference was observed in mean PDT between passages 2 and 8 by late outgrowth EOCs from patients compared with controls (o) (2.2 ± 0.2 vs. 2.3 ± 0.2, p = 0.89) or in CPDL by late outgrowth EOCs from patients compared with controls (p) (22.3 ± 0.7 vs. 22.2 ± 1.4, p = 0.94).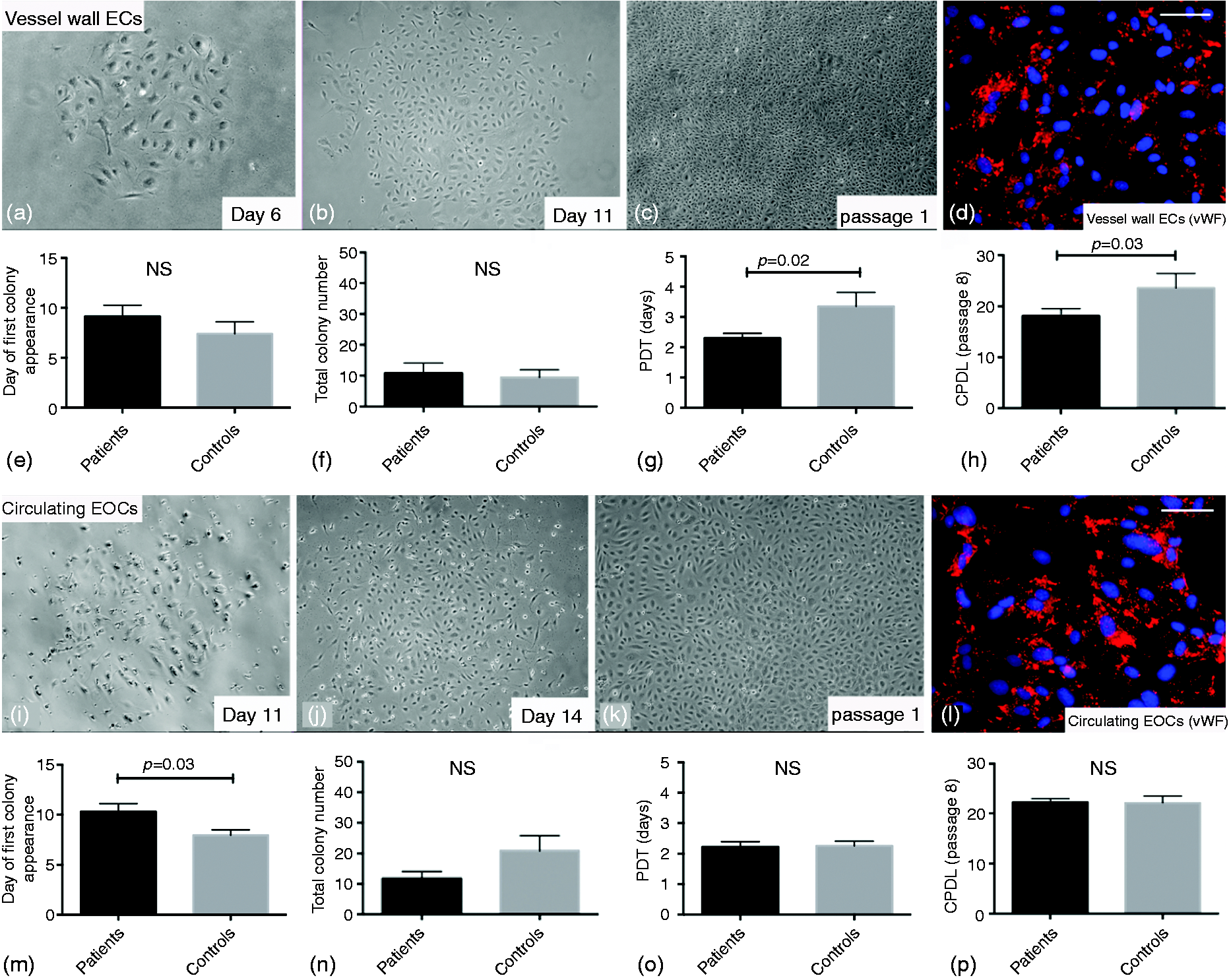
Mean population doubling time (PDT) between passages 2 and 8 by vessel wall endothelial cells from patients was significantly reduced compared with controls (mean PDT ± SEM: 2.3 ± 0.2 vs. 3.4 ± 0.5, p = 0.02) (Figure 2(g)). No significant difference was observed in mean PDT between passages 2 and 8 by late outgrowth EOCs from patients compared with controls (2.2 ± 0.2 vs. 2.3 ± 0.2, p = 0.89) (Figure 2(o)). Similarly, cumulative population doubling levels (CPDLs) of vessel wall endothelial cells from patients were significantly reduced compared with controls (CPDL at passage 8: 18.1 ± 1.4 vs. 23.6 ± 1.4, p = 0.03) (Figure 2(h)). No significant difference was observed in CPDL by late outgrowth EOCs from patients compared with controls (22.3 ± 0.7 vs. 22.2 ± 1.4, p = 0.94) (Figure 2(p)).
Phenotypic characterisation of cells
Vessel wall endothelial cells and late outgrowth EOCs from patients and controls had similar ubiquitous expression of von Willebrand factor (vWF) (Figure 2(d) and (l)). Vessel wall endothelial cells maintained a high expression of mature endothelial cell markers with no differences between patients and controls at early (2–4) passages (CD31 + 98.3 ± 0.6% vs. 96.2 ± 1.4%, p = 0.24; CD146 + 98.5 ± 0.5% vs. 97.5 ± 0.9%, p = 0.15) and late (5–10) passages (CD31 + 97.5 ± 0.6% vs. 97.9 ± 1.2%, p = 0.14; CD146 + 95.6 ± 1.3% vs. 92.9 ± 3.9%, p = 0.89). A relatively high expression of KDR by vessel wall endothelial cells was maintained during culture in patients (89.17Ye haematopoietic antigen, CD45, was expressed at low levels throughout (≤4% for all) (Figure S2 and Table S1 online).
There was no difference in the phenotype of late outgrowth EOCs between patients and controls at early (2–4) passages (CD31 + 96.5 ± 1.0% vs. 92.0 ± 2.9%, p = 0.40; CD146 + 96.6 ± 1.1% vs. 97.4 ± 1.1%, p = 0.26), and late (5–10) passages (CD31 + 94.0 ± 2.5% vs. 96.4 ± 1.4%, p = 0.90; CD146 + 98.8 ± 0.3% vs. 98.3 ± 0.7%, p = 0.80). Expression of KDR by late outgrowth EOCs was similar in early and late passages in patients (71.9 ± 10.1% to 73.8 ± 10.8%, p = 0.45) and controls (70.0 ± 9.1% to 60.1 ± 10.6%, p = 0.47), and did not change significantly in patients compared with controls at late passage (p = 0.14). CD45 expression was low throughout (≤4% for all) (Figure S2 and Table S1 online).
Functional characterisation of cells
In order to identify cell-specific mechanisms that may be dysfunctional and thus contribute to the pathophysiology of premature coronary artery disease, we carried out in vitro functional assessments of migration and adhesion. Vessel wall endothelial cells from patients had reduced migration in a wound-healing assay compared with controls (wound coverage at 24 h: 35.9 ± 8.8% vs. 64.8 ± 12.7%, p = 0.04) (Figure 3(a) to (c)). Migration was similar for late outgrowth EOCs in patients and controls (62.2 ± 8.2% vs. 56.3 ± 8.0%, p = 0.60) (Figure 3(e) to (g)). Cell adhesion was reduced in vessel wall endothelial cells from patients compared with controls (adherent cells per well: 31.5 ± 5.4 vs. 53.3 ± 6.6, p = 0.02) (Figure 3(d)), but there were no differences in adhesion by late outgrowth EOCs (52.0 ± 7.1 vs. 57.3 ± 11.2, p = 0.83) (Figure 3(h)).
Functional analyses of vessel wall endothelial cells and late outgrowth endothelial outgrowth cells. The potential of vessel wall derived endothelial cells (ECs) to migrate and undergo wound healing during early passages (passages 2–4) in vitro was significantly reduced in patients compared with controls (wound coverage at 24 h: 35.9 ± 8.8% vs. 64.8 ± 12.7%, p = 0.04, n = 16 for both) ((a) to (c); × 10 magnification). Conversely, wound healing by late outgrowth endothelial outgrowth cells (EOCs) was similar in patients and controls (wound coverage at 24 h: 62.2 ± 8.2% vs. 56.3 ± 8.0%, p = 0.60, n = 16 for both) ((e) to (g); × 10 magnification). Wound healing assays are representative phase contrast images of endothelial cells ((a) and (b)) and EOCs ((e) and (f)) from a patient with premature coronary artery disease at passage 2. Similarly, cell adhesion by vessel wall endothelial cells was significantly reduced in patients compared with controls (adherent cells per well ± SEM: 31.5 ± 5.4 vs. 53.3 ± 6.6, p = 0.02, n = 16 for both) (d), whereas no differences were observed in adhesion by late outgrowth EOCs in patients and controls (adherent cells per well: 52.0 ± 7.1 vs. 57.3 ± 11.2, p = 0.83, n = 16 for both) (h).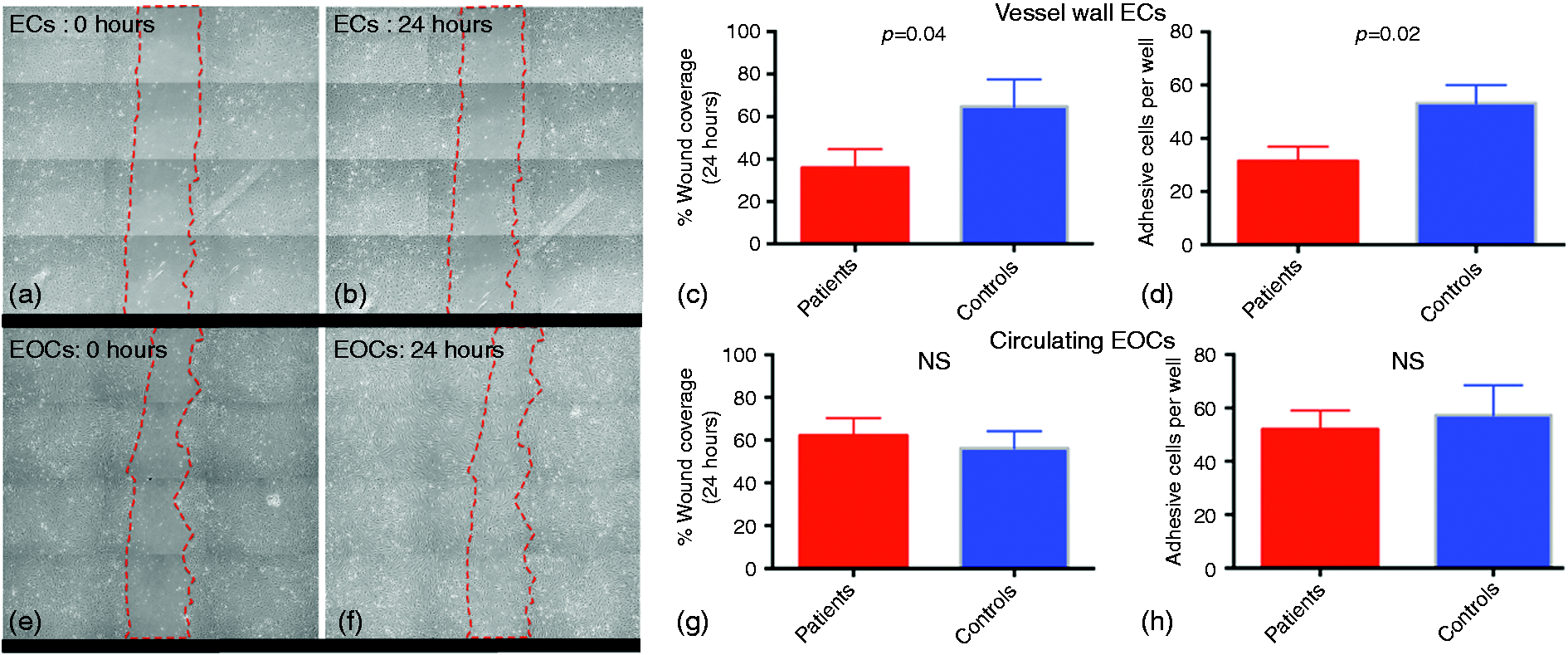
MicroRNA expression by local vessel wall endothelial cells and circulating EOCs
MicroRNA (miR) expression is often selective or enriched in individual cell types and cell specific ‘miR signatures’ can define cellular function. We assessed expression levels of miRs −10 a, −27 b, −let7b, −126 and −181 b in early passage local vessel wall endothelial cells and late outgrowth EOCs from patients and controls (n = 8 for each). Levels of miR−10 a (0.7 ± 0.1 vs. 1.0 ± 0.1, p = 0.007), −let7b (0.64 ± 0.1 vs. 1 ± 0.1, p = 0.0004), −126 (0.69 ± 0.1 vs. 1.0 ± 0.1, p = 0.02) and −181 b (0.69 ± 0.1 vs. 1.0 ± 0.1, p = 0.02) were significantly reduced in vessel wall endothelial cells from patients compared with controls (Figure 4). No differences in miR expression levels were observed in late outgrowth EOCs from patients and controls (p > 0.05 for all) (Figure 4).
Expression of microRNAs known to regulate endothelial function in vessel wall endothelial cells and endothelial outgrowth from circulating cells. Quantitative polymerase chain reaction (qPCR) was used to investigate expression levels of microRNAs (miRs) −10 a, −27 b, −let7b, −126, −181 b in low passage (2–4) vessel wall endothelial cells (ECs) and late outgrowth endothelial outgrowth cells (EOCs) from patients and controls. Relative quantities (RQ) of miR−10 a (0.7 ± 0.1 vs. 1.0 ± 0.1, p = 0.007), −let7b (0.64 ± 0.1 vs. 1 ± 0.1, p = 0.0004), −126 (0.69 ± 0.1 vs. 1.0 ± 0.1, p = 0.02) and -181 b (0.69 ± 0.1 vs. 1.0 ± 0.1, p = 0.02) were significantly reduced in vessel wall ECs from patients compared with controls (n = 8 for each). No difference was observed for the level of miR−27 b in vessel wall ECs in patients compared with controls (0.87 ± 0.2 vs. 1.0 ± 0.2, p = 0.11). For EOCs the levels of miR−10 a (0.88 ± 0.2 vs. 1.0 ± 0.1, p = 0.15), −27 b (1.54 ± 0.6 vs. 1.0 ± 0.2, p = 0.27), −let7b (0.96 ± 0.13 vs. 1.0 ± 0.1, p = 0.28), −126 (0.89 ± 0.2 vs. 1.0 ± 0.1, p = 0.18) and −181 b (0.90 ± 0.2 vs. 1.0 ± 0.1, p = 0.18) were similar in patients and controls (n = 8 for each).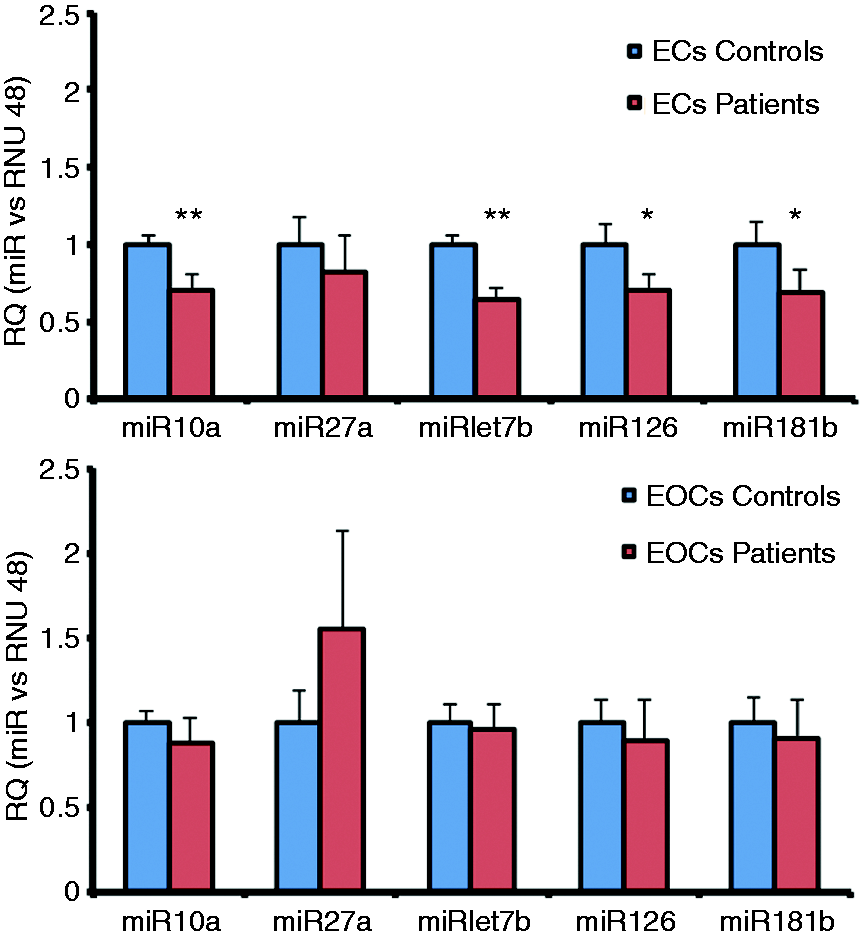
Discussion
The role of local and circulating EPCs in maintaining vascular health is not well understood, with deficiencies in vascular repair potentially contributing to endothelial dysfunction and the development of premature coronary artery disease. We compared numbers of circulating EPCs in patients with premature coronary artery disease and matched controls, and additionally compared endothelial outgrowth following in vitro expansion of peripheral blood MNCs and endothelial cells isolated and expanded from the local blood vessel wall. Whilst patients had endothelial-dependent vasomotor dysfunction compared with controls we found no differences in the number of circulating EPCs or in the function of late outgrowth EOCs isolated from circulating progenitor cells. In contrast, endothelial cells isolated directly from the vessel wall had marked impairment in proliferation, adhesion and migration, and reduced levels of expression of miRs known to regulate endothelial function in patients compared with controls. These findings suggest that impairment of local vessel wall endothelial cells, rather than circulating EPCs, contributes to endothelial dysfunction in patients with premature coronary artery disease.
Previous observations suggest that patients with cardiovascular disease have reduced numbers of circulating EPCs.17–24,26 However, we observed no differences in the number of circulating EPCs or in the function of their progeny in patients and controls. It is important to note that previous reports have defined progenitor cells in blood based on their co-expression of haematopoietic (CD34+, CD133+) and vascular (KDR+) markers. These studies did not study the function of EOCs from these putative EPC populations. We isolated, expanded and characterised EOCs in patients with premature coronary artery disease and demonstrated that these cells are similar in number and function to carefully matched healthy controls. Our findings are consistent with a recent report that EOC proliferation was similar in patients with premature coronary artery disease and their first-degree relatives. 27
Endothelial denudation is one of the earliest pathophysiological features of vascular disease, with persistent endothelial dysfunction responsible for the progression and clinical manifestations of atherothrombosis. 28 The traditional paradigm of vascular repair is based on the proliferation and migration of existing mature endothelial cells from the adjacent vasculature. Our observation that vessel wall endothelial cells have reduced proliferation, adhesion and migration suggest that local mechanisms of vascular repair are impaired in patients who develop premature coronary artery disease. These findings are consistent with previous reports demonstrating endothelial dysfunction in patients with coronary artery disease29,30 and support a key role for endothelial dysfunction in the pathogenesis of coronary artery disease. Whilst the origin of circulating EPCs remains uncertain, the fact that their progeny are not dysfunctional suggests that deficiencies in the number and function of circulating EPCs are not central to the development of premature coronary artery disease.
Why are vessel wall derived endothelial cells but not EOCs from circulating progenitors dysfunctional? We postulated that functional differences might be due to differential expression of miRs and therefore translational repression or degradation of specific messenger RNAs and related transcriptional networks. A number of miRs have been shown to regulate aspects of endothelial function including miR-126,31–38 miR-10 a,34,39 miR−27 b, 40 miR-let7b 41 and miR-181 b.39,42 We demonstrate that expression of these microRNAs was reduced in vessel wall endothelial cells, but not in late outgrowth EOCs in patients with premature coronary artery disease; hence suggesting broad dysregulation of endothelial transcriptional networks. Further studies will explore the pathways through which these microRNAs regulate vessel wall endothelial cell function.
Our findings highlight the importance of studying tissue-appropriate endothelial cells when investigating the pathogenesis of disease.13,16 To date, studies have been challenging in humans due to the challenges associated with accessing vascular endothelial cells. However, we report novel methods for the expansion and maintenance in culture of vessel wall endothelial cells from both patients and healthy controls. This method was adapted from an endothelial biopsy protocol where cells were immediately fixed for quantitative immunofluorescence.43,44 This development permitted the first functional and phenotypic analyses of human vessel wall endothelial cells maintained long-term in culture, and a direct comparison of these cells with circulating EPCs and EOCs from the same patients.
Our study has a number of strengths. First, we identified well-characterised patients and matched healthy controls, and all participants underwent a gold standard assessment of vascular function. Second, we have carefully quantified multiple putative circulating EPC populations, and comprehensively characterised the function of EOCs. Third, we developed novel methods to culture vessel wall endothelial cells from patients to understand the role of local endothelial cells in vascular repair. However, there are also some limitations to consider. We have evaluated EPCs at a single time point in patients with stable disease and it is not possible to determine whether patients have deficiencies in EPC mobilisation at times of acute vascular injury. However, our previous studies suggest that phenotypic EPCs are not mobilised in the context of local vascular injury during angioplasty or stent implantation 6 or systemic injury following acute systemic inflammation. 45 Our patient population was young and selected to have few traditional risk factors for coronary artery disease. Whilst this avoids potential confounding due to conditions such as diabetes mellitus or cigarette smoking that may influence EPC function, our observations may not be generalisable to the broader population of patients with coronary artery disease.
In conclusion, impaired proliferation, adhesion, migration and reduced microRNA expression of local vessel wall endothelial cells, rather than circulating EPCs, was associated with endothelial dysfunction and premature coronary artery disease. Future studies of vessel wall endothelial cells are likely to advance our understanding of the mechanisms of premature coronary artery disease and may lead to novel approaches to enhance vascular repair.
Footnotes
Acknowledgements
We would like to thank the research nurses at the Wellcome Trust Clinical Research Facility, Royal Infirmary of Edinburgh for their invaluable assistance with this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: British Heart Foundation Intermediate Clinical Research Fellowship (FS/10/024/28266), UK British Heart Foundation Cardiovascular Regenerative Medicine Centre Award (RM/13/2/30158).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.