
Abstract
Severe pulmonary hypertension (PH) is rare in chronic obstructive pulmonary disease (COPD). Pulmonary arterial hypertension drugs are vasodilators and may cause severe side effects in these patients. Hence, they are not recommended except in right heart failure on an individual basis. Imatinib, a tyrosine-kinase-inhibitor, has no direct vasodilator effects but significantly improved hemodynamics and exercise capacity in PAH but its use was associated with an increased risk for subdural hematomas in anticoagulated patients. We report on a COPD patient with right heart failure who did not recover with a phosphodiesterase-5-inhibitor or a soluble-guanylate-cyclasestimulator alone but with imatinib as add-on therapy. After one year of treatment, pulmonary vascular resistance (10.8 WU to 2.9 WU), NT-proBNP (4144 pg/mL to 363 pg/mL), and symptoms (WHO FC IV to III, 6MWD bedridden to 303 m) improved without major side effects. Imatinib may be a therapy option in patients with severe PH due to lung disease and right heart failure where other drugs have failed.
Introduction
Although chronic obstructive pulmonary disease (COPD) frequently causes pulmonary hypertension (PH), the degree of hemodynamic impairment is mostly mild.1–4 In a subset of patients, the lung disease is associated with severe PH5–7 and very poor prognosis.6–8 Treatment of PH in severe COPD is difficult because all drugs approved for pulmonary arterial hypertension (PAH) are strong vasodilators that may impair ventilation/perfusion matching. We describe a case of COPD with right heart decompensation due to severe PH, where approved PAH drugs were not sufficient but additional imatinib resulted in an impressive clinical and hemodynamic improvement.
Case report
A 71-year-old male patient with COPD (GOLD 2, Group D) and severe dyspnea (World Health Organization functional class [WHO FC] IV) was admitted to our PH clinic for evaluation and expert opinion. The 6-min walking test could not be performed due to a maximum degree of dyspnea. Pulmonary function test showed moderate obstruction with marked decrease in diffusion capacity for CO (DLCO) (Table 1). He suffered from severe hypoxemia despite continuous high-flow nasal oxygen-therapy. Chest X-Ray (CXR) revealed signs of emphysema, enlarged central pulmonary arteries, and decreased retrosternal airspace. Chest computed tomography (CT) scan showed moderate centrilobular emphysema in the upper lobes and discrete interstitial changes in the basal lung areas but no venous congestion. NT-proBNP was strongly elevated (4144 pg/mL; norm <100 pg/mL). Echocardiography revealed severe right ventricular strain (Table 1). V/Q scan was negative for thromboembolic disease. A right heart catheterization (RHC) confirmed severe precapillary PH (Table 1). As therapy of the lung disease had already been optimized, we started sildenafil at low doses and slowly up-titrated to 20 mg TID. The patient improved slightly and was discharged home. After four weeks, he was admitted to the intensive care unit (ICU) of his local hospital due to right heart failure and after i.v. diuretics he was transferred to our department. Sildenafil was discontinued and the soluble guanylate-cyclase inhibitor (sGCi) riociguat was slowly up-titrated to 2.5 mg TID. This was fairly well tolerated and he was again discharged. After seven weeks he was readmitted to the ICU due to right heart decompensation. After optimization of diuretic management, he was again discharged. At this time point, echocardiography revealed a severely dilated right ventricle (RV; basal diameter = 61 mm) and systolic pulmonary arterial pressure (sPAP) of 88 mmHg. At that time, we offered him a treatment with the tyrosine-kinase inhibitor (TKI) imatinib, starting at a dose of 100 mg BID. The dose was up-titrated to 100 mg QID after two weeks. At the three-month follow-up visit, clinical symptoms improved (6-min walking distance [6MWD] = 303 m, WHO FC III, NT-proBNP = 363 pg/mL). Follow-up echocardiography showed a RV basal diameter of 40 mm and sPAP of 74 mmHg. No significant changes on CXR could be observed. One year after start of imatinib, a follow-up RHC revealed a marked improvement of pulmonary hemodynamics (Table 1). Within the following year, there was no further hospitalization for right heart failure. At the end of this year, imatinib was discontinued for one month due to lack of appetite and mild periorbital edema resulting in improved symptoms but no right heart failure. After restarting imatinib, the adverse effects did not relapse until nine months later, when imatinib was paused for another month. During his last visit in our PH clinic, after 32 months of imatinib, the patient was still in WHO FC III and his last 6MWD was 240 m. He had experienced several COPD exacerbations treated with antibiotics and corticosteroids but no right heart decompensation.
Patient characteristics.
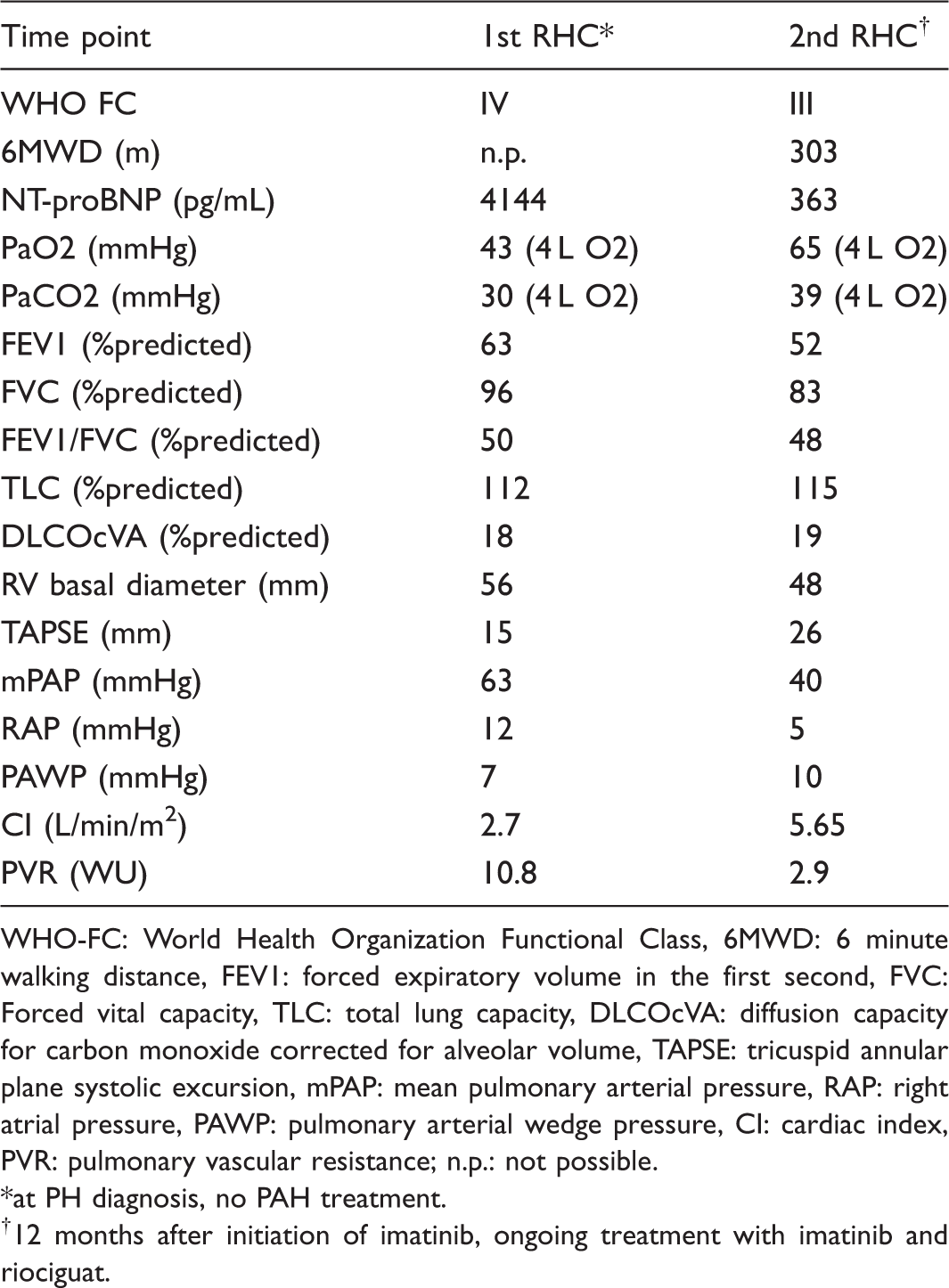
WHO-FC: World Health Organization Functional Class, 6MWD: 6 minute walking distance, FEV1: forced expiratory volume in the first second, FVC: Forced vital capacity, TLC: total lung capacity, DLCOcVA: diffusion capacity for carbon monoxide corrected for alveolar volume, TAPSE: tricuspid annular plane systolic excursion, mPAP: mean pulmonary arterial pressure, RAP: right atrial pressure, PAWP: pulmonary arterial wedge pressure, CI: cardiac index, PVR: pulmonary vascular resistance; n.p.: not possible. *at PH diagnosis, no PAH treatment.
12 months after initiation of imatinib, ongoing treatment with imatinib and riociguat.
Discussion
In patients with severe PH-Lung, it may be difficult to differentiate to which extent PH is caused by the underlying condition or the pulmonary vascular disease. Severe PH-Lung in COPD may also be considered as a pulmonary vascular phenotype of COPD. 4 Some patients with severe PH-Lung and impaired RV-function (mean pulmonary arterial pressure [mPAP] > 35 mmHg or mPAP ≥ 25 mmHg and cardiac index < 2.5 L/min/m2) may benefit from PAH medication.5,9 although there is no evidence from large controlled prospective multicenter studies.
Initial treatment with sildenafil failed to stabilize the patient. We chose sildenafil based on recent studies where this drug showed favorable intrapulmonary selectivity causing beneficial effects on arterial blood oxygenation.10–12 Unfortunately the improvement with sildenafil was not sufficient to stabilize the patient. Riociguat, a sGC stimulator, has strong vasodilative properties that may be superior to the effects of PDE5 inhibitors. 13 However, the combination with PDE5i is contraindicated. This was the rationale for switching from PDE5i to riociguat which also showed beneficial effects in the smoking mouse model. 14 Unfortunately, our patient, despite some clinical benefit, again did not achieve clinical stabilization. Therefore, we decided to offer compassionate treatment with imatinib. Imatinib has no acute vasodilative properties and, therefore, it is not prone to cause ventilation/perfusion mismatch. 15 By inhibition of the PDGF receptor in pulmonary smooth muscle cells it has been shown to reverse pulmonary vascular remodeling in animal models for PH, subsequently leading to normalization of pulmonary hemodynamics and reversion of RV hypertrophy. 16 IMPRES, a randomized controlled double-blind prospective phase III study, had shown that imatinib improved 6MWD, symptoms, and hemodynamics in patients with very severe PAH despite pre-treatment with combined PAH medication. 17 However, the open-label extension study of IMPRES revealed an increased number of subdural hematoma among study patients on anticoagulation mostly with phenprocoumarol in the INR range >2.0. 18 In our patient, there was no indication for anticoagulation, and therefore, the risk of subdural hematoma appeared acceptable, while the risk for severe clinical deterioration without additional treatment was very high.
To our knowledge this is the first report presenting an impressive clinical and hemodynamic improvement after initiation of imatinib therapy in a COPD patient with severe PH. Further studies investigating PDGF inhibitors in patients with lung disease and severe PH after cautious risk/benefit evaluation are warranted.
Footnotes
Conflict of interest
Dr. Douschan reports personal fees and non-financial support from Actelion, non-financial support from Astra Zeneca, non-financial support from Bayer, non-financial support from GSK, non-financial support from MSD, non-financial support from Novartis, non-financial support from Teva, non-financial support from Boehringer Ingelheim, non-financial support from Vifor, non-financial support from Menarini, outside the submitted work. Dr. Kovacs reports personal fees and non-financial support from Actelion, personal fees and non-financial support from Bayer, personal fees and non-financial support from GSK, personal fees and non-financial support from MSD, personal fees and non-financial support from Pfizer, personal fees and non-financial support from AOP, personal fees and non-financial support from Boehringer Ingelheim, personal fees and non-financial support from Novartis, personal fees and non-financial support from Chiesi, outside the submitted work. Dr. Foris reports non-financial support from Actelion, non-financial support from Astra Zeneca, non-financial support from Bayer, non-financial support from GSK, non-financial support from MSD, non-financial support from Novartis, non-financial support from Teva, non-financial support from Boehringer Ingelheim, outside the submitted work. Maria Kuehnelt-Leddihn has nothing to declare. Dr. Olschewski reports grants from Bayer, Unither Pharmaceuticals, Actelion Pharmaceuticals Ltd., Roche, Boehringer Ingelheim, Inventiva and Pfizer Inc., personal fees from Gilead Sciences Inc., Encysive Pharmaceuticals Ltd. and Nebu-Tec, personal fees and non-financial support from Bayer, Unither Pharmaceuticals, Actelion Pharmaceuticals Ltd., Pfizer Inc., Eli Lilly, Novartis, Astra Zeneca, Boehringer Ingelheim, Chiesi, Menarini, MSD and GSK outside the submitted work.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.