
Abstract
Pulmonary arterial hypertension (PAH) is a life-threatening disease characterized by a progressive increase in pulmonary vascular resistance, ultimately leading to right heart failure and death. Throughout the past 20 years, numerous specific pharmacologic agents, including phosphodiesterase-5 inhibitors, endothelin receptor antagonists, prostaglandins, and more recently, soluble guanylate cyclase stimulators and selective IP prostacyclin receptor agonist, have emerged for the treatment of PAH. Early clinical trials were typically of short-term duration, comparing the effects of PAH-targeted therapies versus placebo and using exercise tolerance as the primary endpoint in most trials. A meta-analysis of these trials documented a reduction in short-term mortality of ∼40% with monotherapy. More recently, we have witnessed a progressive shift in PAH study designs using longer event-driven trials comparing the effects of upfront and sequential combination therapy on clinical worsening that is perceived as a more clinically relevant outcome measure. Recent meta-analyses also documented that combination therapy significantly reduced the risk of clinical worsening by ∼35% compared with monotherapy alone.
In this review article, we will discuss the evolution of treatments and clinical trial design in the field of PAH over the past decades with a special focus on combination therapy and its current role in the management of PAH. We will also detail unresolved questions regarding the future of PAH patients’ care and the challenges of future clinical trials.
Keywords
Introduction
Pulmonary arterial hypertension (PAH, group 1 of the clinical classification) is characterized by intense pulmonary vascular remodeling, resulting in increased pulmonary vascular resistance (PVR) and ultimately leading to right heart failure and death.1,2 PAH encompasses a variety of different pathologies, such as idiopathic, heritable, anorexigen-induced PAH, as well as PAH associated with concomitant diseases such as connective tissue disease, congenital heart disease, and portal hypertension. Initially shrouded with mystery, the definition of this orphan disease has greatly evolved in past decades. Even though the pathophysiology of the disease is still incompletely understood, extensive research in the field has led to the identification of three key pathways of abnormal vasoconstriction and cell growth, and the elaboration of numerous specific pharmacologic agents targeting the endothelin, the nitric oxide, and prostaglandin pathways that have been progressively used in combination. This article reviews the evolution of treatments and clinical trial conception in the field of PAH over the past decades with a special focus on combination therapy and its current role in the management of PAH. Unresolved questions regarding the future of PAH patients’ care and the challenges of future clinical trials are also discussed.
Initial days of PAH therapies
The past decades have been thriving in terms of treatments for PAH. However, the field experienced a slow start as it took more than 20 years to develop and approve a specific treatment for PAH after the first World Symposium on Pulmonary Hypertension held in 1973. Before the late 1990s, treatments for PAH patients consisted of supportive therapy with diuretics, digoxin, anticoagulants, and oxygen supplementation with or without calcium channel blockers, the latter being now reserved for selected patients who are responsive to acute vasodilators during right heart catheterization. 3 This treatment approach mainly addressed symptoms related to right heart failure and merely altered the devastating course of the disease, with a median survival of 2.8 years. 4
Subsequent developments in the understanding of PAH led to development of numerous specific therapies targeting the well-described pathways characterizing endothelial dysfunction in PAH: the endothelin-1, nitric oxide, and prostacyclin pathways. In 1995, the U.S. Food and Drug Administration (FDA) approved epoprostenol, a parenteral prostaglandin, making it the very first available specific therapy for PAH. The landmark randomized controlled trial (RCT) confirmed that i.v. prostacyclin was associated with improvement of pulmonary hemodynamics and exercise tolerance. Although survival was not the primary endpoint, epoprostenol was associated with a significantly lower mortality at 12 weeks (0% versus 20%, P < 0.002). 5 The following long-term observational studies suggested that these effects persisted over time.6,7 Subsequently, a multitude of other PAH-targeted molecules were formerly assessed in RCTs, approved, and emerged on the market. The current therapeutic arsenal includes phosphodiesterase-5 inhibitors (PDE5i, sildenafil, and tadalafil), endothelin receptor antagonists (ERA, ambrisentan, bosentan, and macitentan), prostaglandins (epoprostenol, iloprost, treprostinil), and, more recently, the soluble guanylate cyclase stimulator (riociguat) and selective IP prostacyclin receptor agonist (selexipag). Although the primary outcome in initial trials was most commonly the change in six-minute walk distance (6MWD), a meta-analysis of the 23 short-term (mean duration of 14 weeks) RCTs comparing monotherapy with supportive care documented a 43% and 61% reduction in mortality and hospitalization, respectively. 8 Patients in the active arm group also experienced improvements in pulmonary hemodynamics, exercise capacity, and functional status, and health-related quality of life. 9
The journey to combination therapy
Despite these promising findings, a significant proportion of PAH patients had unsatisfactory clinical response on monotherapy and long-term prognosis remained poor, with a mortality rate of ∼15% per year in incident PAH patients.10,11 Combination therapy emerged as a logical alternative to monotherapy. The strategy of combining multiple drugs is not restricted to the field of PAH and has been used extensively in other chronic debilitating diseases such as systemic hypertension, 12 chronic heart failure, 13 and diabetes,14–17 where patients on combination therapy have better outcomes than those on single drug therapy. The rationale is that targeting simultaneously multiple pathways involved in the disease’s pathogenesis rather than increasing doses is expected to lead to additive or even synergistic beneficial effects, further improving patients’ outcomes while minimizing potential drug interactions or adverse events. Moreover, because of the early benefit observed in short-term trials, 8 the principle of equipoise for placebo-controlled trials was considered to be no longer respected, making combination therapy trials a mandatory step to assess novel therapies. As a result, most RCTs performed in the last decade in PAH patients included at least one subgroup of patients on background therapy.
A progressive shift in the clinical trial paradigm
Many of the early clinical trials documented improvement in functional capacity, exercise capacity, and pulmonary hemodynamics with combination therapy compared to monotherapy,18–27 while others failed to demonstrate a significant improvement in their primary endpoint.28–30 In 2011, adding to the already conflicting literature, findings from a systematic review and meta-analysis suggested that combining PAH-targeted therapies did not offer any advantage over monotherapy except a modest increase in exercise capacity. 31 Importantly, clinical trials conducted at that time were mainly of short-term duration and used the 6MWD as the primary endpoint.18–21,26,28 Although baseline 6MWD has a good discriminative capacity to predict outcomes in patients at the time of diagnosis, 32 changes in 6MWD appeared to be an inappropriate surrogate marker of disease progression, and meta-analyses suggested that changes in exercise capacity may not predict clinically relevant events such as all-cause death, hospitalization, or lung transplantation.33–35 There was also the concern of a certain “ceiling” effect in patients who have already been stabilized on background monotherapy, leaving little room for improvement when another therapy was added. 36 As expected, combination therapy trials were shown to result in lower changes in 6MWD 37 compared with monotherapy trials. 8
Directly assessing mortality would have been the most relevant endpoint in PAH trials, but it was felt hardly realizable for a disease like PAH. Indeed, PAH is a rare disease and mortality incidence is low in clinical trials, thus jeopardizing study power and feasibility. Moreover, none of the biomarkers currently used in PAH has been validated as a surrogate clinical endpoint.
38
Consequently, there was dire a need to develop endpoints that would capture clinically relevant events.4,10,11,39,40 Following the example of other chronic diseases, especially chronic left heart failure, the concept of clinical worsening emerged in an attempt to represent events that are undesirable and clinically relevant for patients, including hospitalization, symptomatic progression of disease, treatment escalation, transplantation, atrial septostomy, and death.
41
Although this definition slightly diverged from one study to the other, clinical worsening was proven to effectively predict subsequent mortality events in an observational study from the REVEAL registry,
42
and was shown to be consistently reproducible when adjudicated by an independent committee.
41
We have thus witnessed a progressive change in the design of clinical trials (Fig. 1), from smaller scale short-term trials using clinical worsening as a secondary endpoint to large-scale event-driven trials evaluating combination therapy versus monotherapy of PAH-targeted drugs in which clinical worsening was the primary efficacy endpoint.
Recent paradigm shift in the design of clinical trials in pulmonary arterial hypertension. 6MWD, six-minute walking distance; WHO FC, World Health Organization functional class; PAH, pulmonary arterial hypertension; RCT, randomized control trial.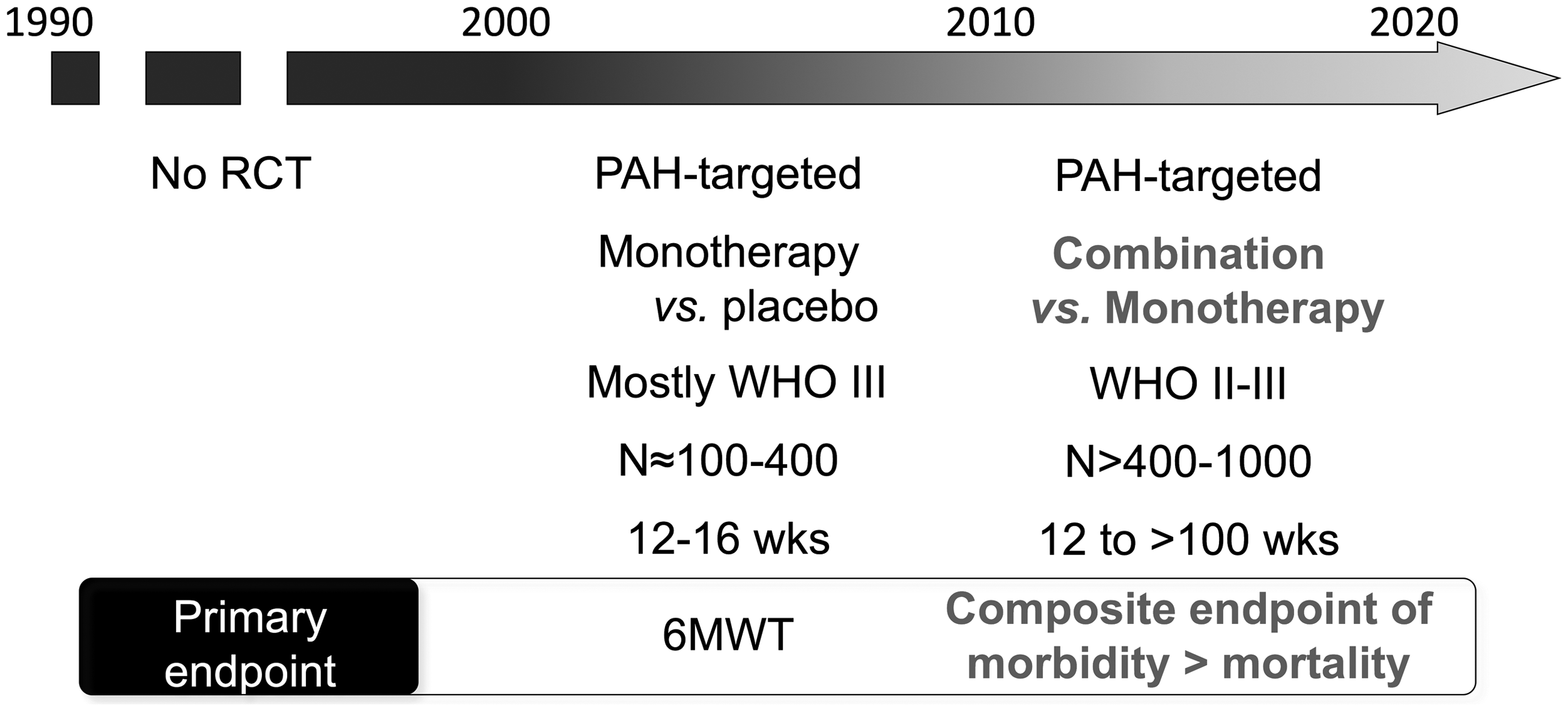
Sequential dual combination therapy
Randomized controlled trials evaluating combination therapy in PAH.

Definitions of clinical worsening varied between studies (see Table 4). *The definition of clinical worsening differed from one study to the other. §The mean duration of study treatment was 63.7 and 70.7 weeks for the patients receiving placebo and selexipag, respectively. ¶80% of patients were on background therapy. ±The mean duration of study treatment was 39.7 months (SD 22.6) and 38.0 months (21.9) for the patients receiving placebo and bosentan, respectively. ≠Only patients on background therapy and randomly assigned to placebo versus macitentan 10 mg (approved dose) per day (308/742, 42%). ≍The mean duration of study treatment was 85.3 weeks and 103.9 weeks for the patients receiving placebo and macitentan 10 mg dose, respectively. ∞Only patients on background therapy and randomly assigned to placebo versus the riociguat 2.5 mg dose groups (191/443, 43%). °Patients pretreated with background therapy only.
Modified intention-to-treat.
6MWD, six-minute walking distance; APAH, associated pulmonary arterial hypertension; CI, cardiac index; CI, confidence interval; CT, combination therapy; CTD, connective tissue disease; CW, clinical worsening; ERA, endothelin receptor antagonist; HR, hazard ratio; HRQoL, health-related quality of life; IPAH, idiopathic pulmonary arterial hypertension; i.v., intravenous; MT, monotherapy; mPAP, mean pulmonary arterial pressure; mRAP, mean right atrial pressure; NT-proBNP, N-terminal-brain natriuretic peptide; PAH, pulmonary arterial hypertension; PDE5i, phosphodiesterase type 5 inhibitors; PVR, pulmonary vascular resistance; SAP, systolic arterial pressure; TTCW, time to clinical worsening; WHO FC, World Health Organization functional class.
Prostanoids in addition to ERAs and/or PDE-5 inhibitors
Seven trials specifically assessed the efficacy of adding non-parenteral prostaglandins to background ERAs and/or PDE-5 inhibitors. In the STEP trial, inhaled iloprost added to background bosentan was not associated with a significant increase in 6MWD (+26 m (P = 0.051), 19 but was associated with improvements in functional status, pulmonary hemodynamics and time to clinical worsening (P = 0.022). The COMBI trial that had a very similar study design and population failed to show any benefit with this combination. 18 Disappointing results were also seen with the addition of oral treprostinil to ERAs, PDE-5i, or both.29,30 Conversely, inhaled treprostinil in addition to background therapy with ERAs or PDE-5i improved exercise capacity. 20 More recently, the GRIPHON study, a multicenter, double-blind, event-driven, phase III RCT confirmed that the addition of selexipag was associated with a 40% decrease in the risk of clinical worsening when compared with placebo in 1156 PAH patients on background therapy. Subgroups of patients receiving either an ERA, a PDE-5 inhibitor, or a combination of the two, corresponding to 15%, 32%, and 33% of the study population, respectively, experienced similar treatment benefit. 44
ERAs in addition to PDE-5 inhibitors or prostanoids
SERAPHIN, a multicenter, double-blind, randomized, event-driven, phase III study randomized 742 patients to receive macitentan 10 mg, macitentan 3 mg, or placebo. In the overall study population, macitentan 10 mg significantly reduced the risk of morbidity/mortality events by 45% (P < 0.001) versus placebo. Predefined subgroup analyses showed that macitentan similarly delayed time to first PAH-related events in both treatment-naïve and pre-treated patients (PDE-5i or non-parenteral prostaglandins), confirming that sequential combination therapy improves long-term outcomes in PAH. 45 Macitentan also improved functional status and pulmonary hemodynamics. 46 More recently, however, the COMPASS-2 study, a double-blind, placebo-controlled, phase IV clinical trial, where 334 patients on baseline sildenafil were randomized to bosentan or placebo, failed to demonstrate a reduction in the risk of morbidity/mortality. An exploratory analysis suggested that bosentan on top of sildenafil improved 6MWD at week 16 (+22 m, P = 0.01). 47 Whether the discrepancy between these two trials is related to differences in the study population and design, power of the study, drug efficacy, or drug–drug interactions (bosentan reducing plasma levels of sildenafil and tadalafil) remains unknown. Importantly, the COMPASS-2 trial had an important amount of missing data due to premature discontinuation of study before any event of clinical worsening. Also, the trial was designed to detect a treatment effect of 40% with combination therapy and the events encountered in the trial were too few. Thus, the study may have lacked statistical power to detect a smaller difference between treatments.
PDE-5i or soluble guanylate cyclase stimulation in addition to prostanoids or ERAs
In the PACES-1 study, 21 the addition of sildenafil in patients with poor exercise capacity despite background i.v. epoprostenol was associated with significant delay in clinical worsening compared to placebo. Improvements in exercise capacity (adjusted treatment difference of 29 m, P = 0.001) and hemodynamics were also observed. Although not a predefined endpoint, more deaths were recorded in the placebo group compared with the sildenafil group. Riociguat also improved exercise capacity after 12 weeks in both treatment-naïve patients and those on background PAH therapy (mostly ERAs). 27 Conversely, the addition of tadalafil on top of bosentan did not result in significant improvement in exercise capacity (+23 m, P = 0.09). 26 The last two trials were not designed to assess the effect of combination therapy with riociguat or tadalafil on clinical worsening.22,23
Initial upfront combination therapy
Only two RCTs compared initial upfront combination therapy versus monotherapy in treatment-naïve patients. In the BREATHE-2 trial, upfront combination of epoprostenol and bosentan was associated with a −36 ± 4% decrease of total pulmonary resistance from baseline to week 16, compared with a 23 ± 3% decrease in the epoprostenol/placebo group (P = 0.08 for the difference between groups). 28 Unfortunately, with a small number of patients (n = 33), it lacked power to detect significant difference between treatments. 28 More recently, the AMBITION trial 48 has brought new evidence in favor of upfront combination therapy in treatment-naïve patients. In this event-driven, double-blind, placebo-controlled RCT, 500 patients with WHO functional class II–III were randomized into three treatment arms: ambrisentan and tadalafil in upfront combination or monotherapy of ambrisentan or tadalafil combined with placebo. Upfront combination therapy led to a 50% reduction in clinical failure compared with the combined monotherapy arms (HR = 0.50, 95% CI = 0.35–0.72, P < 0.001). 48 Importantly, this benefit was not influenced by baseline patients’ characteristics, precluding the identification of the small subgroup of patients that has an excellent long-term prognosis on monotherapy. Patients on combination therapy also had better performance on the 6MWD (median increase of 49 m versus 24 m, P < 0.001) and larger decrease of NTproBNP (–67% versus −50%, P < 0.001). Although adverse events were more common in the combination therapy group, there was no difference among groups for serious adverse events and treatment discontinuation. More recently, a post-hoc analysis of the AMBITION study evaluating survival at seven days after the termination of each individual patient’s randomized treatment suggested a lower mortality in patients initially treated with combination therapy (1% versus 4%, HR = 0.21, 95% CI = 0.06–0.73, P = 0.0065). 49 Because of the exploratory nature of this analysis, this hypothesis needs to be confirmed in future studies. Finally, a recent retrospective analysis of real-world data suggested that these effects were similar regardless of the combination regimen used (ambrisentan/bosentan and tadalafil/sildenafil), 50 which also remains to be confirmed.
Overall assessment of combination therapy efficacy and safety
Secondary outcomes of combination therapy vs. monotherapy in PAH.
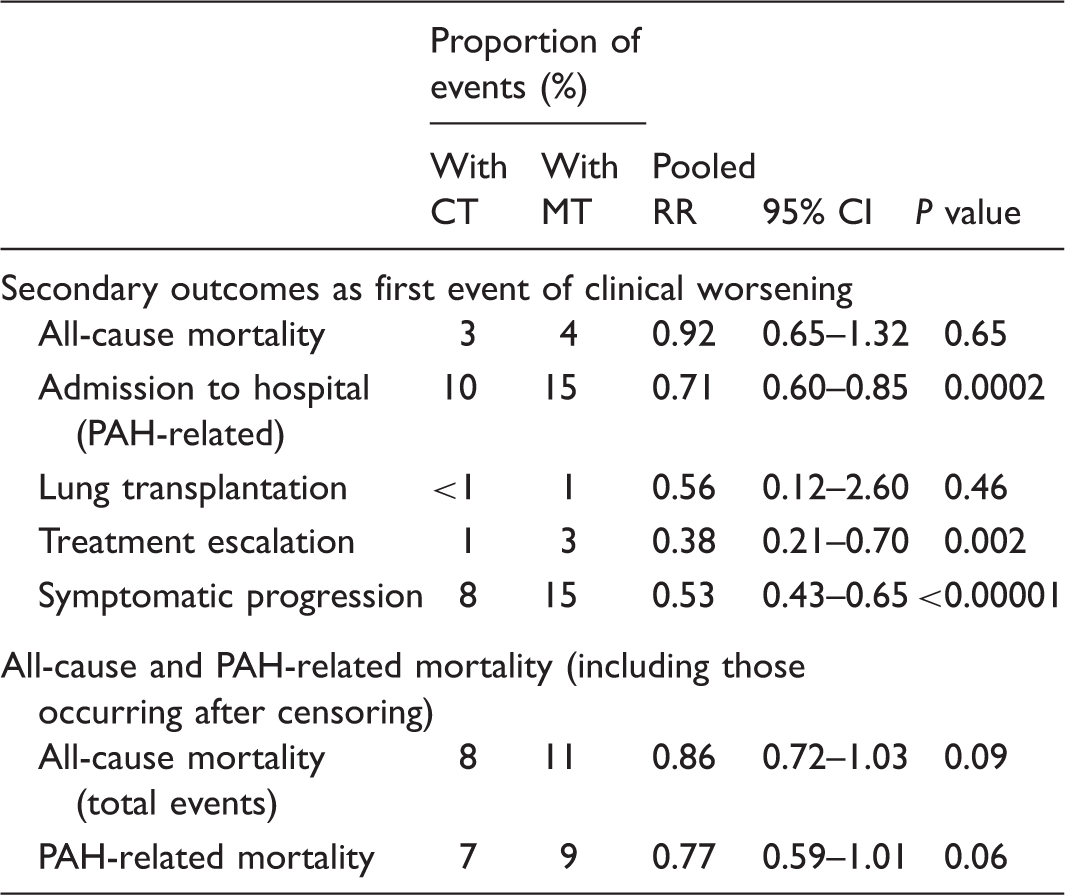
Adapted from Lajoie et al. 37
CI, confidence interval; CT, combination therapy; MT, monotherapy; PAH, pulmonary arterial hypertension; RR, risk ratio.
Predefined subgroup analyses for the risk of clinical worsening with combination therapy compared with monotherapy.
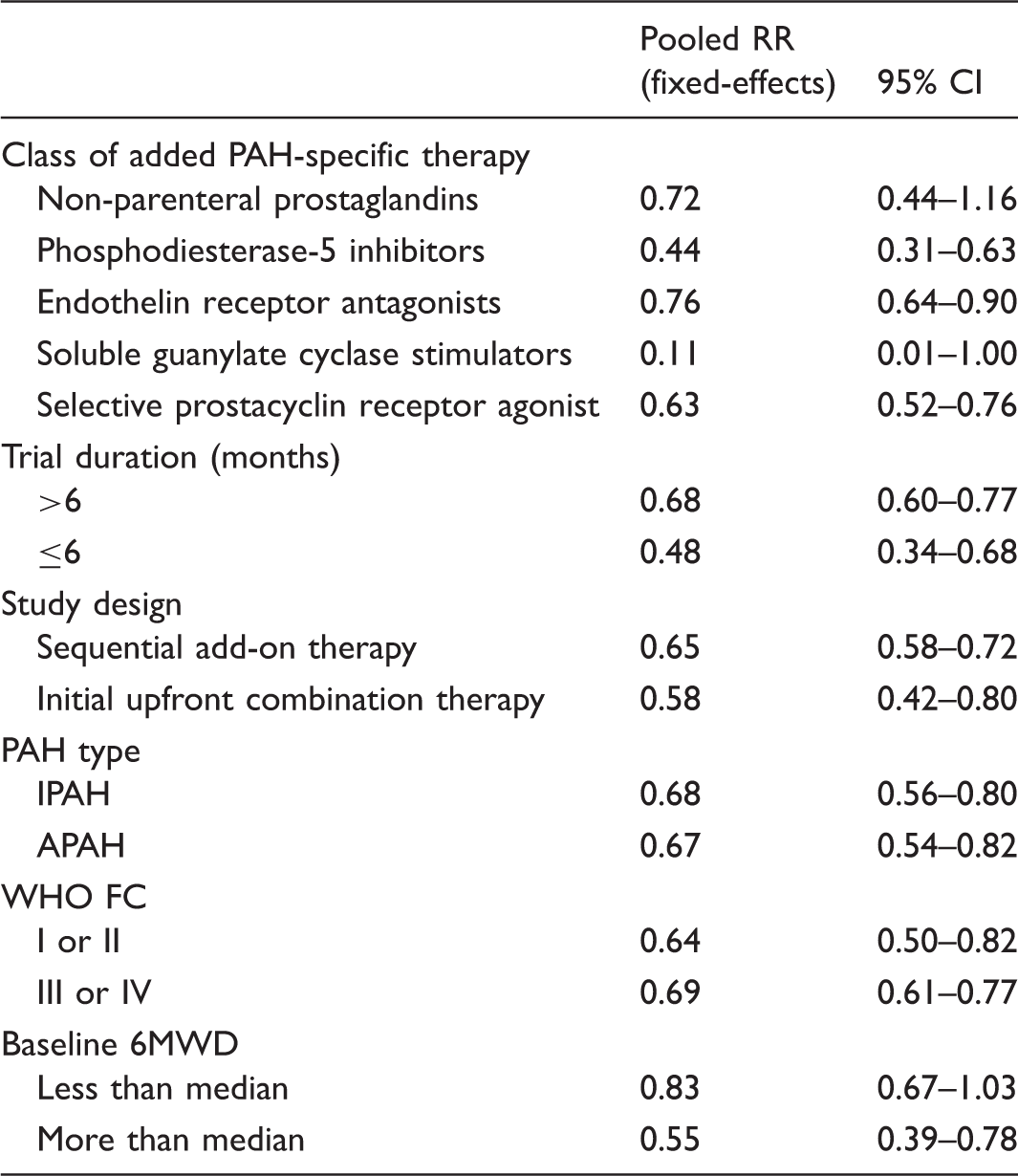
Adapted from Lajoie et al. 37
6MWD, six-minute walking distance; APAH, associated pulmonary arterial hypertension; CI, confidence interval; IPAH, idiopathic pulmonary arterial hypertension; PAH, pulmonary arterial hypertension; RR, risk ratio; WHO, World Health Organization functional class.
Unsolved questions
Although a growing body of evidence confirms that combination therapy of PAH-targeted drugs delays progression of disease, there are still many clinically relevant but unsolved questions.
Upfront versus sequential combination therapy
Sequential combination therapy is the most widely used strategy, both in clinical practice and clinical trials.18–24,26,27,29,30,44,45,47 It consists of adding a second, or perhaps third, drug to background therapy in order to achieve satisfactory clinical response according to a goal-directed approach. This treatment strategy was found to be effective in improving patients’ prognosis in observational studies. 51 Current guidelines2,52 also recommend that therapy should be increased until patients reach a WHO-FC I or II or a near normalization of resting cardiac index or NT-proBNP plasma levels. 53
Until recently, the body of evidence supporting upfront combination therapy was scarce. As stated earlier, the recent AMBITION trial convincingly demonstrated that upfront combination therapy with ambrisentan and tadalafil reduced the risk of clinical worsening compared with initial monotherapy. 48 It is noteworthy, however, that the two drugs were initiated at half of their maximum approved doses and then up-titrated to their maximum approved doses (tadalafil 40 mg and ambrisentan 10 mg) over a period of eight weeks. Thus, it remains unknown whether upfront combination therapy improves long-term outcomes compared with rapid (e.g. after three to four months) sequential add-on therapy in case of unsatisfactory response to initial monotherapy such as persistence of WHO FC ≥ 3, low exercise capacity, elevated brain natriuretic peptide, and poor hemodymamics (right atrial pressure ≥8 mmHg or cardiac index <2.5 L/min/m2), as recommended. 2 As stated above, it is also unclear at this time if these results represent a class effect or are specific to these two agents.
Choice of combination therapies
In the past, because they were fewer oral formulations, combination therapy most commonly consisted of a combination of an ERA and a PDE5i until parenteral prostaglandins were required. The recent arrival of new orally delivered drugs, such as soluble guanylate cyclase stimulators or selective IP prostacyclin receptor agonists, has increased the choice of combination therapy. Importantly, no information exists on the optimal drug combination. Recently, the concept of network meta-analysis emerged as an alternative way to indirectly compare PAH-targeted therapies. An extensive network meta-analysis including 31 studies and over 6000 patients treated with current PAH-specific therapies alone or in combination suggested that ERA, PDE5i, riociguat, and a combination of ERA/PDE5i significantly reduced clinical worsening and improved functional capacity. 54 Surprisingly, they concluded that only the ERA/PDE5i combination was associated with reduced hospitalization. Even though they were associated with the most important increase in functional capacity, parenteral prostaglandins were associated with higher adverse events and treatment discontinuation, which is also consistent with findings of standard meta-analyses.37,43
It is noteworthy that while these meta-analyses strengthen the benefits of combination therapy on clinical worsening in PAH, they are not meant to assess the best treatment strategy. Indeed, comparisons among treatments would be only indirect and subject to artefacts caused by study designs and duration, patient populations, and other co-variables, and should therefore be interpreted with extreme caution in the absence of head-to-head clinical trials. Moreover, these results are not necessarily generalizable to all molecules within the same class of drugs. Indeed, individual therapies differ in terms of pharmacokinetics, pharmacodynamics, selectivity, and drug–drug interactions, and only approximately half of the theoretically possible dual combinations have been systematically evaluated in RCTs. Therefore, the choice of the initial PAH therapy is dependent on a variety of factors including PAH type, approval status, route of administration, patients’ preference, side effect profile, drug–drug interactions, physicians’ experience, and disease severity.
Dual versus triple combination therapy
Despite the reduction in clinical worsening with dual combination therapy, many patients still witness events of clinical deterioration or fail to reach established treatment goals. Triple combination therapy therefore seems the logical next therapeutic option, although current evidences supporting sequential or upfront triple combination therapy are scarce. Nowadays, no RCTs were specifically designed to assess the benefits of sequentially adding a third molecule to dual combination therapy in PAH. Nonetheless, the study population of some clinical trials, such as the FREEDOM-C, FREEDOM-C2, and GRIPHON studies, have included 32–45% of patients on background combination therapy.29,30,44 Interestingly, a prespecified analysis of the GRIPHON study confirmed that triple combination therapy with selexipag still reduced morbidity/mortality events by 37% compared with dual combination therapy of ERA/PDE5i. 44
Upfront combination triple therapy was evaluated in one retrospective pilot study where 19 patients with newly diagnosed, severe PAH were initiated on upfront triple combination therapy sildenafil, bosentan, and i.v. epoprostenol. 55 After four months, they observed significant improvements in 6MWD and a 67% decrease in PVR. Overall survival estimates were better than expected survival calculated from the French equation (100% versus 49% at three years, respectively). 55 This offers preliminary evidence that upfront triple combination therapy could be beneficial in patients with severe PAH at presentation. The ongoing TRITON study, a phase 3b trial, will attempt confirming the role of upfront combination therapy by comparing upfront triple therapy of macitentan, tadalafil, and selexipag versus a combination of macitentan and tadalafil plus placebo on PVR (primary endpoint). 56
Cost-effectiveness of combination therapy
It is well established that the treatment of chronic diseases is a burden on the financial health of a society and great efforts have been made to try reducing the costs related to the treatment of these diseases. Previous studies confirmed that the economic burden of PAH is substantial, with direct healthcare costs per patient per month in the range of $2576–$11,875 (excluding indirect cost).57–59 However, few studies have evaluated the cost-effectiveness of PAH drugs. 60 Cost-effectiveness is frequently assessed by estimating the incremental cost per quality-adjusted life-years (QALYs), with values generally less than US$50,000 but up to US$200,000 being considered cost-effective. 61 A recent cost-effectiveness analysis suggested that first-line monotherapy was associated with cost higher than CAN$140,000 and CAN$350,000 for PDE5i and ERA, respectively. It is noteworthy, however, that although many clinical trials reported measured quality of life, 9 none have reported the impact of treatment in a format that would allow precise estimation of QALYs. These calculations generally rely on utility estimates based on WHO FC that are derived from a single cohort 62 and few studies evaluated the cost-effectiveness of combination therapy. 63 More importantly, the available economic evaluation studies are likely country-specific and have merely concentrated on certain PAH treatments, and the lack of head-to-head comparison studies covering all comparable medications limit the validity cost-effectiveness comparisons. Nonetheless, the emergence of novel drug therapies will necessitate the assessment of their cost-effectiveness, especially when it comes to combination therapy. Further studies addressing this issue and weighting against the efficacy, tolerability, availability, and patient preference will thus be needed.
Challenges of future clinical trials in PAH
The tremendous advances in the treatment of PAH over the last few years are indisputable. However, recent meta-analyses confirmed that patients on combination therapy still witness a significant number of clinical worsening events, which is probably an underestimation of the real disease burden since most patients included in PAH trials were prevalent cases with a substantial proportion of WHO FC I–II. In addition to answering unresolved questions, future clinical trials will face numerous challenges.
Study definitions of clinical worsening in combination therapy trials.
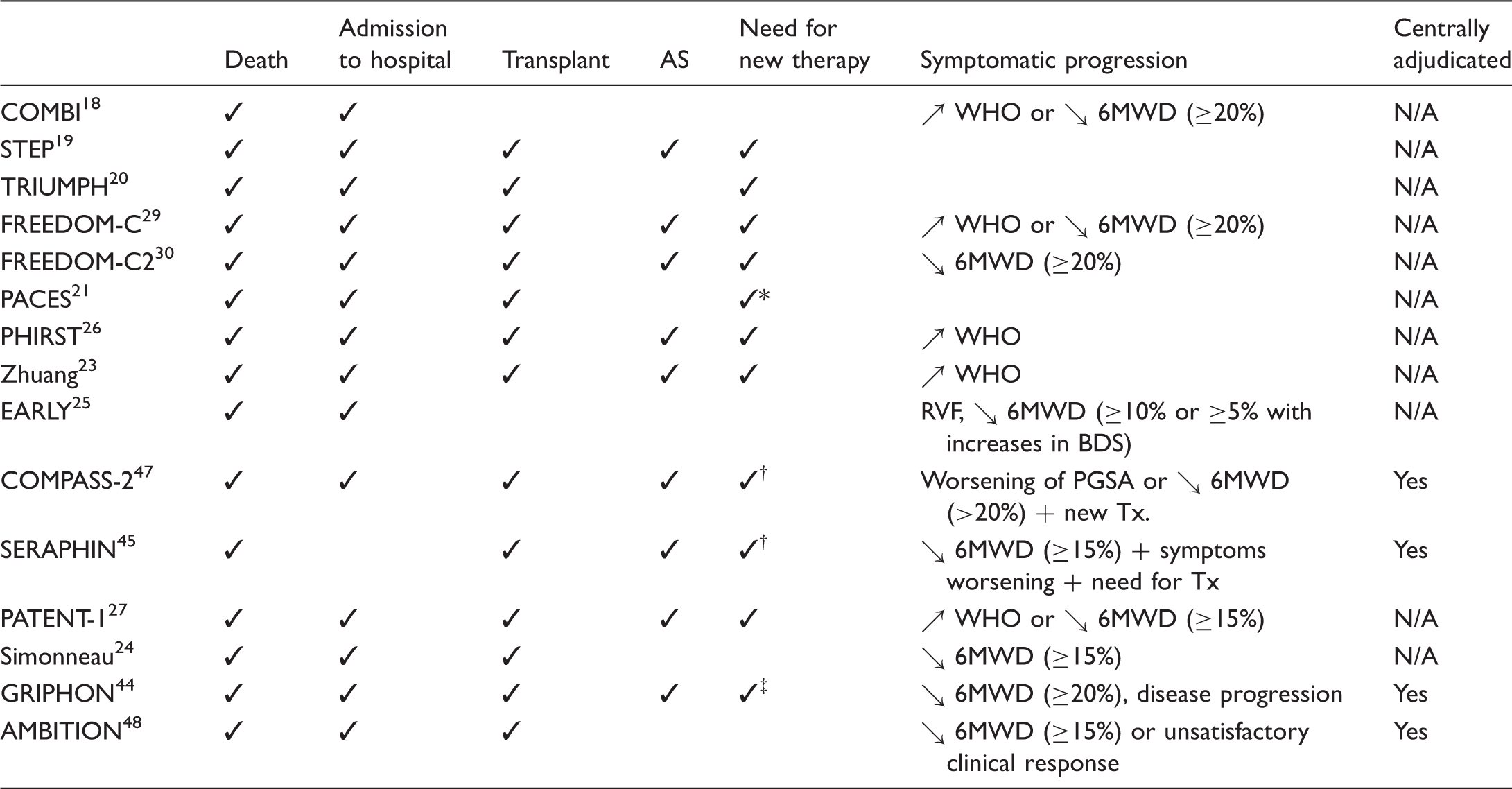
Initiation of bosentan or ↗ in epoprostenol dose (>10%).
Initiation of parenteral prostaglandin.
Initiation of parenteral prostaglandin or LTOT.
6MWD, six-minute walking distance; AS, atrial septation; BDS, Borg dyspnea scale; N/A, not applicable; PGSA, patient global self-assessment scale; RVF, right ventricular failure; WHO, World Health Organization.
Future clinical trials will also need to address the problem of informative censoring inherent to the time to clinical worsening endpoint. Often in such trials, only the first event of clinical worsening is reported, subsequent events being ignored. As an example, a recent meta-analysis noted that PAH-related mortality was reported for fewer than 50% of patients contributing to the mortality assessment. When all deaths (first event or not) were taken into account, they observed a trend toward mortality reduction. 37 Thus the time to first event might have underestimated the true impact of therapy on mortality. It is important to keep in mind that the treatment effect for the composite outcome is not necessarily the same as the effects on its individual components. Therefore, future studies should be designed to provide enough power not only to detect a clinically relevant effect for the composite, but also detect an impact on its individual components.
In the attempt to better capture events of clinical worsening, clinical trials also evolved from short-term trials with a fixed length to long-term trials continued until a prespecified number of clinical events occurred. These recent event-driven studies lasted four to six years, patients being exposed to the study drugs on average for approximately two years.44,45,47,48 However, the treatment effect was evident by 12 months. There are thus certain ethical considerations in maintaining a patient in a study for such lengthy periods. In the context of an orphan disease with limited and competing recruitment for trials and the rapidly changing treatment paradigm in PAH, the optimal duration of future trials should be revisited, balancing study power with the possibility for patients to contribute to subsequent trials and benefit from newer PAH-target therapies and treatment algorithms.
It has also become increasingly difficult for clinical studies to obtain the statistical power required to detect a reduction in clinically relevant endpoints, especially mortality, since there are few events in the populations studied. Populations currently being studied are most commonly prevalent rather than incident PAH patients, and a significant proportion of them have a relatively preserved FC. In order to improve statistical power in clinical trials, enriching the population of patients enrolled in these studies has been proposed as a possible solution. Previous observational studies and registries4,11 as well as a recent sub-study of the SERAPHIN trial confirmed that incident PAH patients have an increased rate of clinical worsening despite comparable baseline characteristics. 64 Therefore, enrolling more patient with recent PAH diagnosis would be one way to enrich patient population in PAH clinical trials. Another alternative trial would be to include a greater proportion of patients who are clinically deteriorating since it is known that patients who recently had an event of clinical worsening are at higher risk of mortality. 42 This is a concept largely used in clinical trials in heart failure and idiopathic pulmonary fibrosis.65,66 These suggestions do not address, however, the real need to move forward to endpoints that reflect disease improvement rather than progression as a relevant and important goal for PAH patients.
Novel clinical trial designs are also increasingly being used in other chronic diseases. 67 The N of one study design systematically evaluates different treatments in the same patient. The patient is thus exposed to a predetermined treatment for a predetermined period of time after which he is exposed either to placebo or another active therapy. This allows a more personalized approach, but makes the results difficult to generalize to an entire population. The factorial design allows testing multiple hypotheses at once and can be conducted as a 2 × 2 confrontation where treatment A and treatment B are matched with placebos and combined in different fashion. However, interaction between drugs must be taken into account when conducting such a trial.
Finally, albeit great effort and promising results have been made to delay disease progression in PAH with combination therapy, it remains an ultimately incurable disease.37,43 We are entering an exciting era for new therapeutics in the treatment of PAH when novel agents are expected to synergize with currently approved vasodilators to reverse vascular remodeling. 68 Novel potential targets of PAH drug currently under development target vascular inflammation, autoimmunity, metabolic derangements, and aberrant BMPRII signaling.69,70 These newer trials also face specific challenges. First, these new approaches will need to demonstrate benefit on top of currently available therapies68,71 a barrier not faced in the original PAH trials. This makes the detection of any putative benefit potentially challenging. Moreover, some new therapies carry novel risks of adverse events not encountered with approved agents, including immunosuppression, epigenetics,72,73 and metabolism. 74 Finally, the pathway to development for these novel drugs may not be supported by standard industry, requiring convincing-enough rationale to site investigators who are accustomed to getting more attractive compensation for examining already-trusted vasodilation pathways.
Conclusion
There is a growing body of evidence confirming that dual combination of PAH-targeted therapy significantly delays disease progression in PAH patients. Combination therapy has progressively become the standard of care treatment for a large proportion of patients with advanced PAH. However, patients’ quality of life and long-term prognosis remain suboptimal for many of them. Future research is thus mandatory to identifying the best treatment strategy, such as initial upfront versus rapid sequential combination and dual versus triple combination therapy, as well as to investigating treatments beyond the traditional signaling pathways targeted by the currently available PAH therapies.
Footnotes
Conflict of interest
ACL has no conflict of interest; SB holds a Canadian Research Chair in translational research in pulmonary vascular diseases at Université Laval; SP is clinician-scientist of the Fonds de Recherche en Santé du Québec and has received research grants from Actelion Pharmaceuticals, Bayer and GlaxoSmithKline, and has received speaker fees from Actelion Pharmaceuticals. The Pulmonary Hypertension Research Group is also supported by the “Réseau en Santé Respiratoire” of the FRSQ.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.