
Abstract
We have demonstrated that simvastatin and sphingosine 1−phosphate (S1P) both attenuate increased vascular permeability in preclinical models of acute respiratory distress syndrome. However, the underlying mechanisms remain unclear. As Krüppel-like factor 2 (KLF2) serves as a critical regulator for cellular stress response in endothelial cells (EC), we hypothesized that simvastatin enhances endothelial barrier function via increasing expression of the barrier-promoting S1P receptor, S1PR1, via a KLF2-dependent mechanism. S1PR1 luciferase reporter promoter activity in human lung artery EC (HPAEC) was tested after simvastatin (5 μM), and S1PR1 and KLF2 protein expression detected by immunoblotting. In vivo, transcription and expression of S1PR1 and KLF2 in mice lungs were detected by microarray profiling and immunoblotting after exposure to simvastatin (10 mg/kg). Endothelial barrier function was measured by trans-endothelial electrical resistance with the S1PR1 agonist FTY720-(S)-phosphonate. Both S1PR1 and KLF2 gene expression (mRNA, protein) were significantly increased by simvastatin in vitro and in vivo. S1PR1 promoter activity was significantly increased by simvastatin (P < 0.05), which was significantly attenuated by KLF2 silencing (siRNA). Simvastatin induced KLF2 recruitment to the S1PR1 promoter, and consequently, significantly augmented the effects of the S1PR1 agonist on EC barrier enhancement (P < 0.05), which was significantly attenuated by KLF2 silencing (P < 0.05). These results suggest that simvastatin upregulates S1PR1 transcription and expression via the transcription factor KLF2, and consequently augments the effects of S1PR1 agonists on preserving vascular barrier integrity. These results may lead to novel combinatorial therapeutic strategies for lung inflammatory syndromes.
Keywords
Introduction
The pathophysiology of sepsis, 1 acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), 2 hypertension, 3 atherosclerosis,4,5 and ischemia-reperfusion injury 6 is commonly characterized by loss of vascular integrity and accumulation of plasma-borne proteins and tissue edema. Importantly, therapeutic strategies that inhibit loss of endothelial barrier function have been associated with strategies that improve outcomes. 7
ARDS is a life-threatening condition characterized by profound inflammation, increased vascular permeability, and alveolar flooding, leading to severe hypoxemia, respiratory failure, and >30% mortality.8–10 Despite advances in care of the critically ill, there is an urgent need to improve our understanding of the biochemical/genetic basis of ARDS in order to broaden the limited therapeutic options currently available.
The statins are a class of HMG-CoA reductase inhibitors with potent anti-inflammatory and vascular-protective properties independent of their serum cholesterol lowering capacity. 11 We and others have shown simvastatin to improve human endothelial cell barrier function both in vitro and in vivo 5 with attenuation of murine ARDS by pretreatment with simvastatin. 12 In humans, simvastatin attenuates the pulmonary and systemic inflammatory response to intravenous LPS in health 13 and patients. 14 Simvastatin reduced LPS-induced BAL neutrophilia, TNFα, matrix metalloproteinases, and C-reactive protein. 15 The mechanisms underlying these effects, however, are complex and not fully understood.
In prior work, we also identified the sphingosine 1–phosphate receptor 1 (S1PR1) to be critically involved in the regulation of lung vascular permeability and in the development and progression of ARDS. 16 S1PR1 regulates multiple aspects of the inflammatory response, including cell proliferation, vascular barrier regulation, and leukocyte diapedesis by binding to its natural ligand, sphingosine 1-phosphate (S1P).17–20 S1PR1–/– mice exhibited marked vascular barrier disruption and embryonic hemorrhage leading to intrauterine death. 21
Similar to S1PR1, the transcription factor, Krüppel-like factor 2 (KLF2), also known as lung Krüppel-like factor, is highly expressed in vascular endothelial cells. KLF2−/− embryos expire from severe hemorrhaging with endothelial cell necrosis. 22 KLF2 is a key transcriptional regulator of the vasoprotective effects of shear stress 23 and transcriptional regulator of endothelial proinflammatory activation; 24 KLF2 acts as a central transcriptional switch point between the quiescent and activated states of the adult endothelial cell 25 and p53 impairs endothelial function by transcriptionally repressing KLF2. 26
We hypothesize that statins enhance S1PR1 expression and signaling via increased transcription induced by KLF2 to attenuate lung inflammation and ARDS. In the present study, we studied the interactions of simvastatin, KLF2, and S1PR1 expression to explore the mechanisms of statin-mediated endothelium protection in ARDS.
Materials and methods
Cell culture and reagents
Human pulmonary artery endothelial cells (ECs) were obtained from Lonza (Walkersville, MD, USA), and cultured as previously described18,27 in EBM-2 Complete Medium (Cambrex) at 37℃ in a humidified atmosphere of 5% CO2 and 95% air, with passages 6–10 used for experiments. Unless otherwise specified, reagents were obtained from Sigma (St. Louis, MO, USA). ON-TARGETplus siRNA for KLF2 (siKLF2) and control siRNAs (siControl) were purchased from Dharmacon (Lafayette, CO, USA). pCMV6-KLF2 and pCMV6-XL4 were purchased from OriGene Technologies (Rockville, MD, USA). Rabbit anti-S1PR1 antibodies were purchased from Exalpha Biologicals (Watertown, MA, USA). Rabbit anti-KLF2, anti-β-actin antibody and LPS were purchased from Sigma (St. Louis, MO, USA). Secondary horseradish peroxidase (HRP)-labeled antibodies were purchased from Amersham Biosciences (Piscataway, NJ, USA).
Animal preparation and experimental intervention
Male C57BL/6 J mice (age 8–10 weeks; Jackson Laboratories, Bar Harbor, ME, USA) were administered 10 mg/kg simvastatin (Sigma, St. Louis, MO, USA) or vehicle by intraperitoneal injection three times a week for six weeks. The last dose was given 24 h before the sample collection. Mice were anesthetized with intraperitoneal ketamine (100 mg/kg) and acetylpromazine (1.5 mg/kg), and mice lungs were subsequently collected for mRNA and protein, as we previously described.12,28–30
RNA isolation and microarray analysis
As we previously described,12,28–30 total RNA was extracted from lungs using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and was used to synthesize double-stranded cDNA. Biotin-labeled antisense cRNA was then generated and hybridized to the Affymetrix Mouse Genome 430 2.0 Array, as described in the Affymetrix GeneChip protocol (Affymetrix, Santa Clara, CA, USA). Arrays were normalized and processed using Bioconductor GCRMA package. Gene expression profile of S1PR1 and KLF2 were derived from the array 29 and their expression in the simvastatin and control groups were further analyzed and compared. These microarray datasets have been shared on NCBI-GEO (accession: GSE14431)
Immunoblotting
Cellular materials from treated or untreated ECs, or homogenized mice lungs, were incubated with RIPA buffer (Cell signaling Co., Danvers, MA, USA) as we previously described. 30 Solubilized proteins were separated by SDS-PAGE in 4–15% polyacrylamide gels, transferred to Immobilon membranes. After blocking non-specific sites with 5% BSA, the blots were incubated with either rabbit anti-S1PR1 antibody or anti-KLF2 antibody, followed by incubation with HRP-labeled goat anti-rabbit IgG. The visualization of immunoreactive bands was achieved using enhanced chemiluminescence (ECL).
Subcloning, transfection, and luciferase activity assays
DNA fragments of 2 kb human S1PR1 gene promoter were amplified by polymerase chain reaction (PCR) using the human genomic DNA as previously described. 31 The fragments were fused to a pGL3-basic reporter vector (Promega, Madison, WI, USA) and transfected into ECs. A plasmid with renilla luciferase gene (phRL-TK) was co-transfected by Fugene HD (Promega Co., Fitchburg, WI, USA) as a control. siKLF2 or siControl, pCMV6-KLF2 or pCMV-XL4 were transfected to EC for testing the functions of KLF2. Transfected EC were cultured in growth medium, exposed to simvastatin 5 μM for 12 h and lysed in passive lysis buffer. Luciferase activity was measured by Dual-Luciferase Assay Kits and GloMax-Multi Detection System (Promega). The relative activities were expressed as the ratio of firefly luciferase in pGL3 to renilla luciferase in phRL-TK (RLU). Six independent transfections and duplicate luciferase assays were performed for each condition.
Chromatin immunoprecipitation (ChIP) and PCR
To investigate the interaction between S1PR1 promoter and transcription factor KLF2, ChIP assay was performed with EZ-Magna ChIP A/G kit (EMD Millipore, Billerica, MA, USA) in accordance with the manufacturer’s procedure. Briefly, EC cells were fixed by 1% formaldehyde, lyzed with SDS lysis buffer, and sheared with three 15-s pulses in a VirSonic 60 sonicator (SP Scientific/VirTis, Warminster, PA, USA); then the supernatant containing protein/DNA complexes was incubated with KLF2 antibody and protein A magnetic beads overnight at 4℃. After washing, elution, crosslink reversal, and DNA purification, PCR of DNA was performed by a CFX384/C1000 thermal cycler (BioRad, Hercules, CA, USA). Four KLF transcription factor-binding sites within S1PR1 core promoter 1 kb from TSS were identified in silico with Genomatix software (www.genomatix.de). Primers were designed to capture the one nearest to TSS using forward-1 (5’– TCAGCACACCGATCCTCCTA) and reverse-2 (5’– GATAGACGCTGATCCCTGGC) which yields a product of 182 base pairs that captures the KLF binding site. PCR was performed using EvaGreen Supermix (BioRad, Hercules, CA, USA) as per the manufacturer’s protocol.
Measurement of EC electrical resistance
ECs were grown to confluence in polycarbonate wells containing evaporated gold microelectrodes. ECs were stimulated by S1PR1 agonist FTY-(S)-phosphonate (Tys) 500 nM and trans-endothelial electrical resistance (TER) measurements were performed using an electrical cell substrate impedance sensing system obtained from Applied Biophysics (Troy, NY, USA) as described in details. 27 TER values from each microelectrode were pooled at discrete time points and plotted versus time, and the values of area under curves were calculated and compared.
Statistical analysis
The t-test and one-way ANOVA analysis were used for the comparison of luciferase activities of S1PR1 promoter, mRNA, and protein levels of S1PR1 and KLF2 under different conditions. Statistical significance was defined at P < 0.05 in both tests. All experiments were repeated three times unless indicated otherwise.
Results
Simvastatin significantly increases S1PR1 expression in vitro and in vivo
Utilizing human lung EC transfected with the S1PR1 luciferase reporter, we detected that simvastatin (5 μM, 12 h incubation) significantly increases human S1PR1 promoter activity (two-fold increase) (*P < 0.05) (Fig. 1a). Incubation with simvastatin (5 μM) also significantly increased expression of the endogenous S1PR1 protein (Fig. 1b). Consistent with these in vitro effects, levels of S1PR1 mRNA and protein were significantly increased in lungs from mice after simvastatin exposure (10 mg/kg, i.p., three times/week for six weeks) indicating strong in vivo effects of simvastatin on S1PR1 transcription (*P < 0.05) (Fig. 1c) and protein expression (Fig. 1d).
Simvastatin significantly increased S1PR1 gene transcription and protein expression in vitro and in vivo. (a) Simvastatin increased human S1PR1 promoter activity in human pulmonary artery endothelial cells (HPAEC). Relative luciferase activities of pGL3-S1PR1-promoter to phRL-TK in HPAEC were detected by dual-luciferase reporter assay following simvastatin (5 μM) for 12 h, normalized by ones without simvastatin. S1PR1 promoter activity was increased by simvastatin (about twofold) (n = 4) (*P < 0.05 vs. control). (b) Simvastatin increased S1PR1 protein expression in HPAEC. Endogenous S1PR1 protein expression in HPAEC was measured by immunoblotting with ani-S1PR1 after simvastatin (5 μM) for 16 h, compared with ones without simvastatin, controlled by β-actin protein level (n = 3). (c) Simvastatin increased S1PR1 gene transcription in mice lungs. Alteration of S1PR1 mRNA expression in lungs from mice was detected by microarray after simvastatin 10 mg/kg or vehicle Intra-peritoneal injection (i.p.) three times per week for six weeks (*P < 0.05 vs. control) (n = 3). (d) Simvastatin increased S1PR1 protein expression in mice lungs. Alteration of S1PR1 protein expression in lungs from mice was measured by immunoblotting with anti-S1PR1 after simvastatin 10 mg/kg or vehicle three times per week for six weeks (n = 3).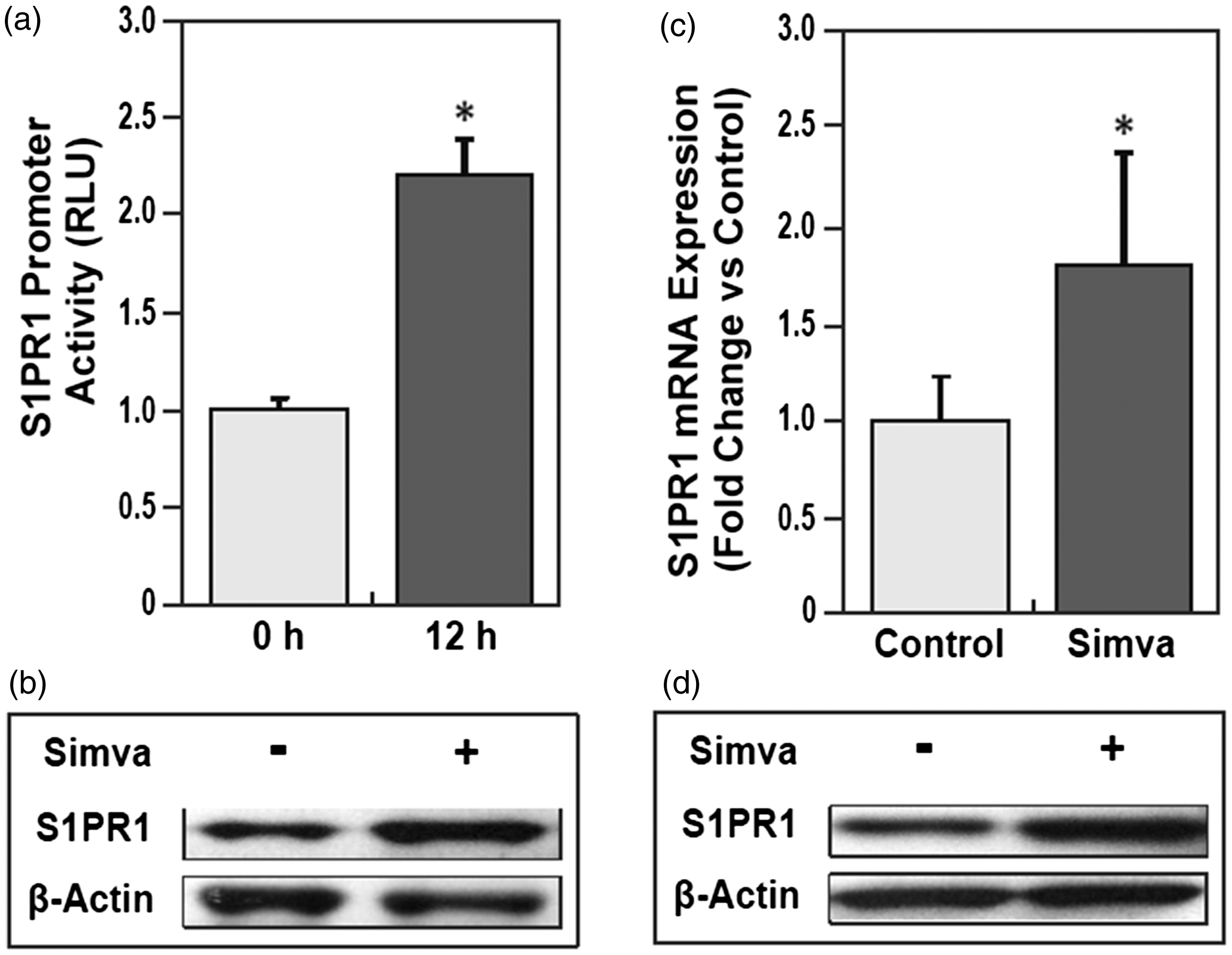
Simvastatin significantly increases KLF2 expression in vitro and in vivo
Krüppel-like factor 2 (KLF2) is a critical regulator for cellular stress response in endothelial cells (EC),
24
and acts as a central transcriptional switch point between the quiescent and activated states of ECs.
25
Also, KLF2 is reported as an important transcriptional regulator of the statin-mediated effects in vascular endothelium.
32
We next evaluated KLF2 expression levels in human lung endothelial cells and mice lungs following simvastatin treatments. Both in vitro and in vivo exposure of either human lung EC to 5 μM simvastatin or mice to 10 mg/kg simvastatin (i.p.) resulted in significantly increased KLF2 protein expression in human lung ECs (Fig. 2a), and significantly increased both KLF2 mRNA transcription (P < 0.05) (Fig. 2b) and protein expression (Fig. 2c) in murine lungs.
Simvastatin significantly increases gene expression of transcription factor KLF2, which positively regulates endogenous S1PR1 protein expression. (a) Simvastatin increased KLF2 protein expression in HPAEC. Endogenous KLF2 protein expression in HPAEC was measured by immunoblotting with ani-KLF2 after simvastatin (5 μM) for 16 h, compared with ones without simvastatin, controlled by β-actin protein level. (b) Simvastatin increased KLF2 gene transcription in mice lungs. Alteration of KLF2 mRNA expression in lungs from mice was detected by microarray after simvastatin 10 mg/kg or vehicle i.p. three times per week for six weeks (*P < 0.05 vs. control). (c) Simvastatin increased KLF2 protein expression in mice lungs. Alteration of KLF2 protein expression in lungs from mice was measured by immunoblotting with anti-KLF2 after simvastatin 10 mg/kg or vehicle i.p. three times per week for six weeks. (d) Over-expression of KLF2 by transfection of pCMV6-KLF2 for 36 h, compared with empty vector pCMV6, significantly increased endogenous S1PR1 protein expression in EC. (e) Reductions in KLF2 expression by transfection of siKLF2 for 72 h, compared with siControl, significantly decreased endogenous S1PR1 protein expression in HPAEC (n = 3).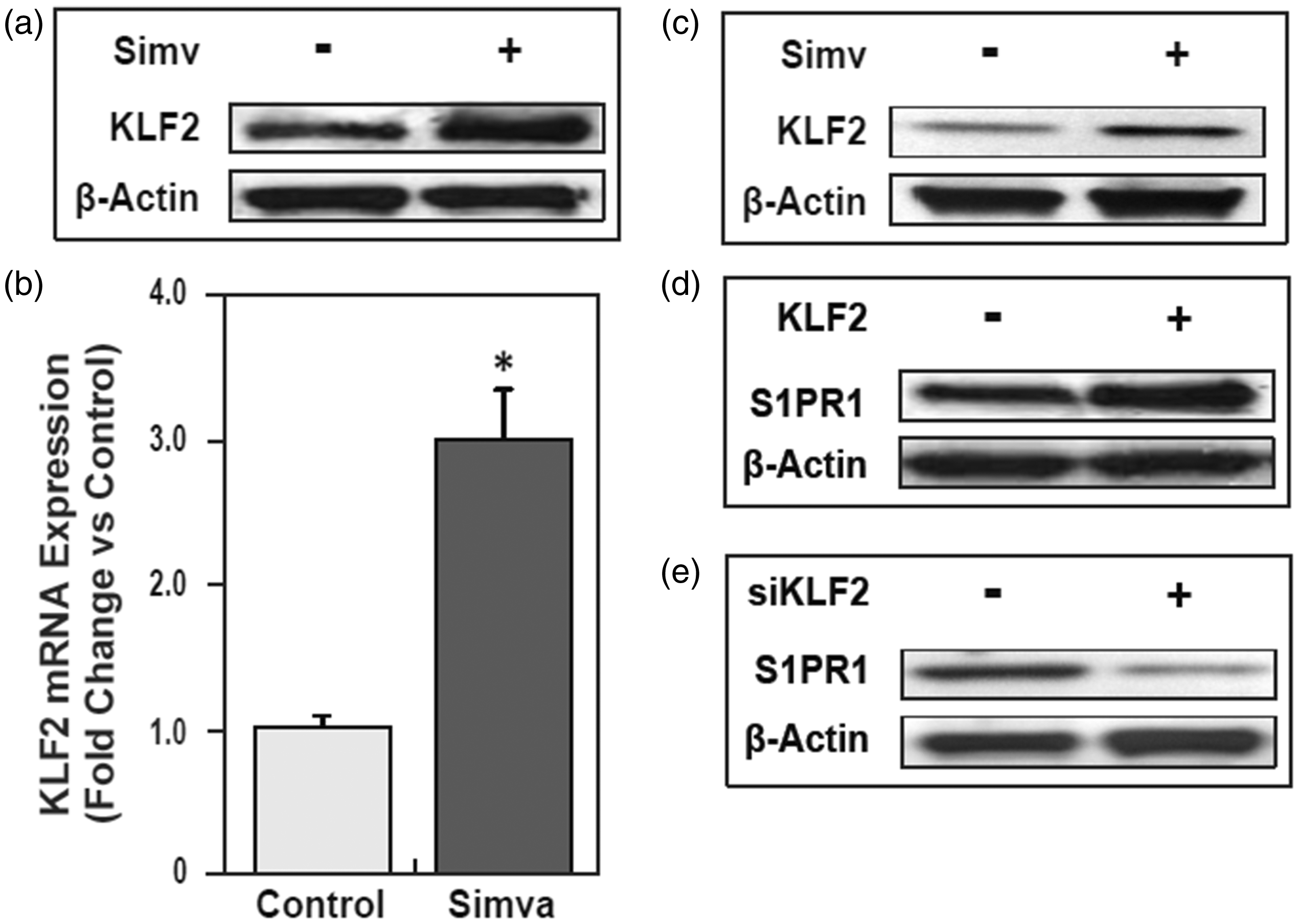
KLF2 significantly increases S1PR1 protein expression
In silico analysis by two software tools, Genomatix (www.genomatix.de) and Jaspar (jaspar.genereg.net), revealed four potential KLF binding sites located in the 1 kb human S1PR1 proximal promoter residing at −864 to −840, −673 to 655, −496 to −473, and −286 to −263 bp from transcription start site (TSS). There are some differences between the DNA sequence of mouse and human S1PR1 promoter, but these two promoters have three highly conserved KLF binding sites in proximal promoter regions. We next tested effects of KLF2 on S1PR1 gene transcription and expression. The results demonstrated that increased KLF2 expression by transfection of pCMV-KLF2 in HPAECs significantly increased endogenous S1PR1 protein expression (Fig. 2d), compared with ones transfected by empty vector. Consistent with these results, reduced KLF2 expression by KLF2 siRNA (siKLF2) resulted in significantly decreased endogenous S1PR1 protein expression compared to siRNA controls (Fig. 2e), again indicating that KLF2 positively regulates S1PR1 protein expression.
Simvastatin significantly enhanced S1PR1 promoter activity via KLF2
To investigate the effects of KLF2 on simvastatin increasing S1PR1 transcription and protein expression, we altered KLF2 expression in HPAECs by siKLF2 (Fig. 3a) and treated EC with simvastatin (5 μM). These results demonstrated that silencing of KLF2 dramatically attenuated simvastatin-induced increase of S1PR1 promoter activity (Fig. 3b), indicating simvastatin significantly increases S1PR1 gene transcription activity via the transcription factor, KLF2.
Simvastatin increased S1PR1 promoter activity by increasing KLF2 recruitment to the S1PR1 promoter, which is significantly attenuated by reduced KLF2 expression. (a) KLF2 protein expression in human lung HPAEC was significantly reduced by transfection of siKLF2 for 72 h comparing to ones by siControl, and increased by simvastatin (5 μM) in both original and induced KLF2 by silencing. (b) Reduced of KLF2 by transfection of siKLF2 significantly attenuated simvastatin (5 μM) mediated increase of S1PR1 promoter activity (*P < 0.05 vs. no siControl; #P < 0.05 vs. siControl/Simva). (c) Human HPAEC cells were exposed to simvastatin (5 μM) or vehicle for 4 h, and fixed by 1% formaldehyde. KLF2 cross-linked protein/DNA complexes were immunoprecipitated (IP) with rabbit anti-KLF2. PCR was performed with primers designed to capture the KLF2-binding site near the transcription start site of the S1PR1 promoter, negatively and positively controlled by IgG and total input without IP.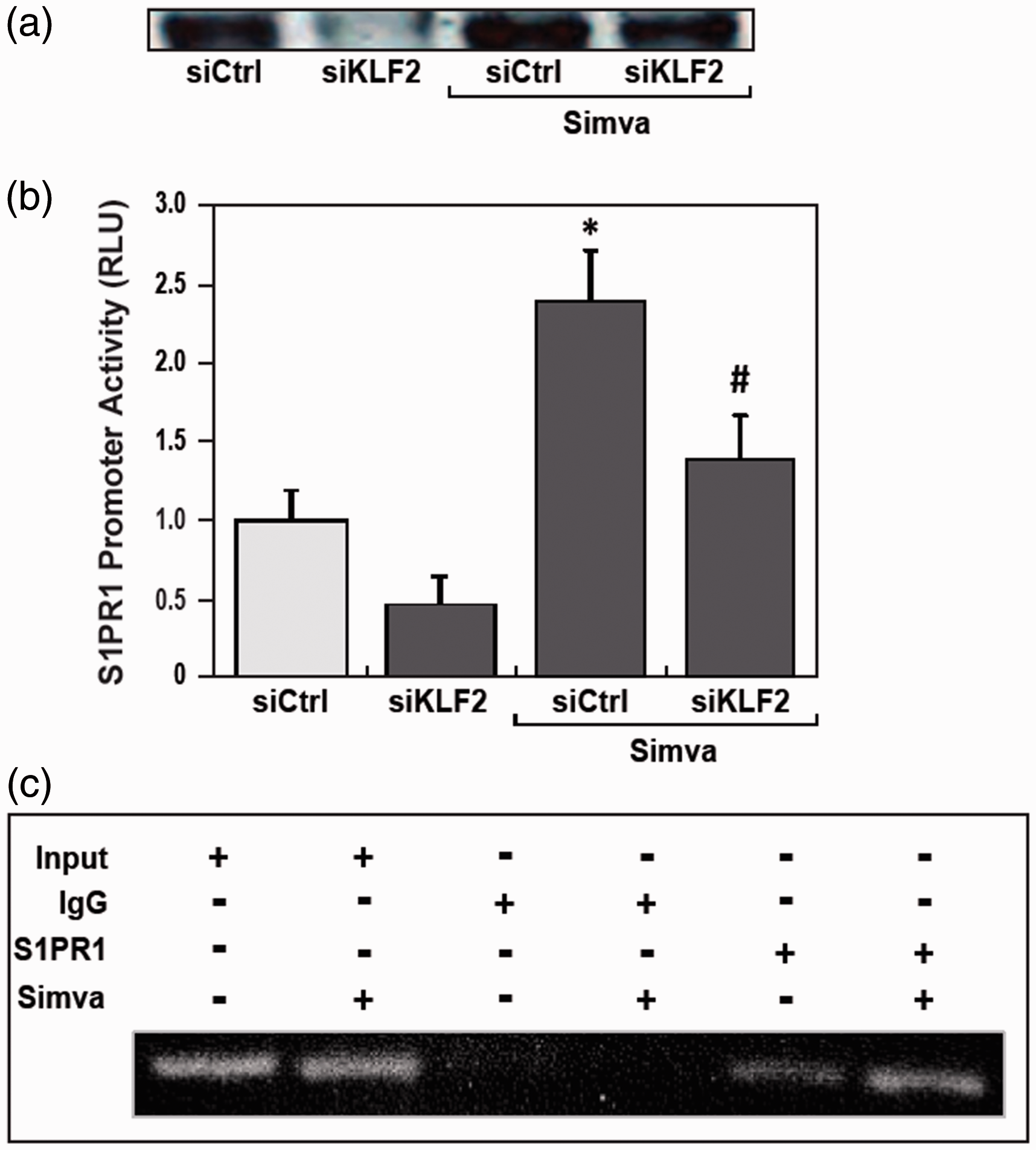
Simvastatin significantly increases KLF2 recruitment to the S1PR1 promoter
In general, transcription factor efficiency on gene promoter activity is highly influenced by the distance from the TSS.33,34 The potential KLF2 binding sites on S1PR1 promoter −286 to −263 bp from TSS, were tested by protein-DNA binding ChIP assay with KLF2 antibodies and PCR with defined primers. Compared with controls, KLF2 protein binding to this S1PR1 promoter region was enhanced by simvastatin treatment (Fig. 3c). These results demonstrate that simvastatin significantly increased KLF2 recruitments to the S1PR1 promoter. It is indicated that simvastatin increased transcription factor KLF2 binding to S1PR1 promoter, and hence increased S1PR1 promoter activity, gene transcription, and protein expression.
Simvastatin significantly augments S1PR1 agonist effects on endothelial barrier enhancement
To reveal the roles of simvastatin in S1PR1 mediated increase of endothelium barrier integrity, we measured trans-endothelial electronic resistance (TER) of human lung EC response to S1PR1 agonist FTY-(S)-phosphonate (Tys) 500 nM with or without simvastatin pre-treatment (Fig. 4a). Simvastatin treatment dramatically enhanced effects of S1PR1 agonist on TER of lung ECs (P < 0.05), which was significantly attenuated by KLF2 silencing (P < 0.05) (Fig. 4b). It is indicated that simvastatin significantly augments the effects of S1PR1 on the endothelium barrier enhancements and the attenuation of vascular permeability.
Simvastatin significantly augments S1PR1-mediated human lung endothelial barrier enhancement. (a) Human lung HPAEC was transfected by siKLF2 or siControl for 72 h, then treated by simvastatin (5 μM) or vehicle (control) for 16 h, trans-endothelial electronic resistance (TER) of human lung EC response to S1PR1 agonist FTY-(S)-phosphonate (Tys) 500 nM was measured by electrical cell substrate impedance sensing system, compared with no Tys and normalized by each baseline before stimulation. (b) TER curves of S1PR1 receptor in HPAEC response to its agonist Tys was analyzed by area under curves (AUC). Compared with controls, simvastatin significantly increased S1PR1 agonist Tys mediated increase of TER of lung ECs (*P < 0.05 vs. Tys only), and siKLF2 significantly attenuated the enhancement effects of simvastatin, compared with siControl (#P < 0.05 vs. Simva/Tys).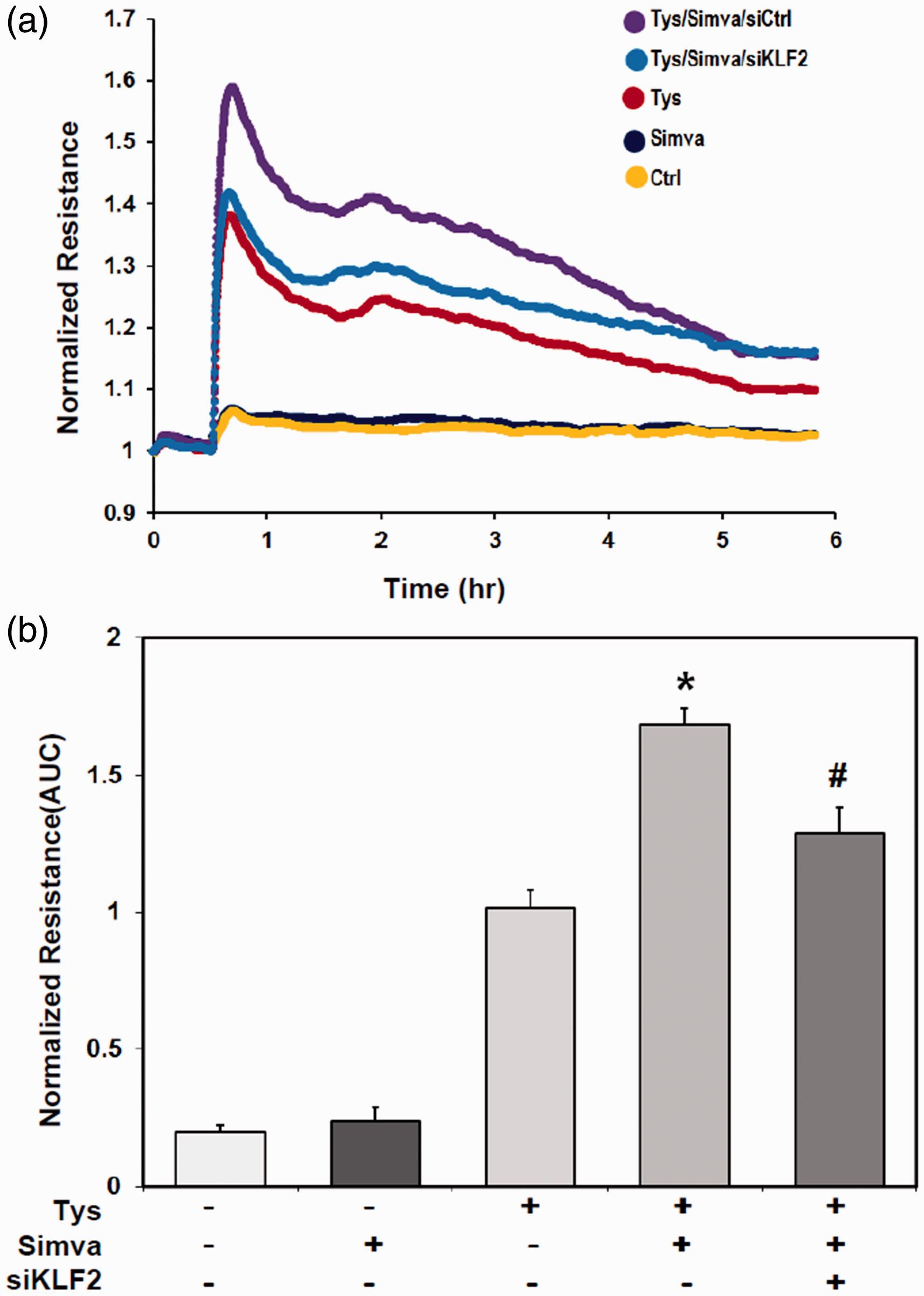
Discussion
In addition to its wide-spread utilization to reduce cholesterol levels, the HMG-CoA reductase inhibitor, simvastatin, is also a potent anti-inflammatory and vascular-protective reagent in murine and human inflammatory diseases, including ARDS.12,14 To further explore mechanistic aspects of the protective effects of simvastatin on human lung endothelial barrier properties, we investigated effects and mechanisms of simvastatin on the gene expression and function of the critical vascular barrier-regulatory receptor, S1PR1.18,27,35
In the present study, we found that simvastatin significantly increased S1PR1 gene expression in vitro and in vivo via lung transcription factor KLF2, and augmented the function of S1PR1 on endothelial barrier enhancement. Utilizing luciferase reporter assays, cDNA microarray, and protein western blotting, we confirmed that simvastatin significantly increased S1PR1 promoter activity, mRNA transcription, protein expression in human lung ECs as well as in murine lung tissues, which were significantly regulated by KLF2. Consistent with these results, simvastatin significantly increased the specific S1PR1 agonist FTY720-(S)-phosphonate (Tys)36-mediated increases in trans-endothelial resistance.
In our previous studies, we found that pretreatment of simvastatin had potent beneficial effects on vascular function via modification of intracellular events that drive cellular contraction, paracellular gap formation. 37 We observed marked attenuation of vascular leak and inflammation by pretreatment of simvastatin in a murine model of endotoxin-induced ALI in association with identifiable histological protection. Whole lung tissue microarray analysis revealed clear differential expression of genes involved in several gene ontologies relevant to ALI (inflammation and immune response, transcription, cell signaling). 12 Previous reports performed in bovine aortic endothelial cells demonstrated that pitavastatin or atorvastatin significantly increased protein and mRNA expression of the S1PR1 receptor but not the S1PR3 receptor. 38 In human aortic smooth muscle cells, simvastatin significantly increased both S1PR1 and S1PR3 transcription. 39
However, the mechanism of statin-mediated S1PR1 upregulation has not been previously addressed. We explored the underlying molecular mechanism of simvastatin regulating S1PR1 expression with a particular focus on KLF2, a key transcription factor and transcriptional regulator of the vaso-protective effects of shear stress, 23 KLF2 is reported to mediate transcriptional events evoked by several statins (simvastatin, lovastatin, mevastatin) including expression of endothelial nitric oxide synthase and thrombomodulin in EC 40 and the atheroprotective effects of C-type natriuretic peptide. 32 KLF2 is also a key regulator of endothelial barrier function mice lung and human vein endothelial cells. 41 In our mice models of hyperventilation- and LPS-induced ALI, both hyperventilation and LPS dramatically depressed gene expression of KLF2 in mice lung by DNA microarray analysis (data not shown). Also, lung KLF2 expression was significantly decreased by paraquat-induced ALI in Wistar rats 42 suggesting that KLF2 expression may be an early indicator of ALI/ARDS.
Our study demonstrates that KLF2 significantly increases S1PR1 protein expression in human lung ECs and murine lungs. In Jurkat T cells, S1PR1 was reported as one of the most highly upregulated genes upon KLF2 induction, with 5.8-fold change, by microarray analysis. 43 Over-expression of KLF2 positively regulates S1PR1 expression by activating its promoter. 44 KLF2 controls thymocyte and T-cell migration by binding to S1PR1 promoter and trans-activating S1PR1 transcription. 45 We demonstrated that in human lung endothelial cells, KLF2 mediated simvastatin-induced S1PR1 expression by increased binding and transactivation of the S1PR1 promoter to contribute the effects of simvastatin.
Simvastatin improves disturbed endothelial barrier function, both dose- and time-dependent, and was accompanied by a reduction in the thrombin-induced formation of stress fibers, 5 which are similar but not to the barrier-regulatory effects of S1P. 27 Our earlier work revealed that S1P, the natural ligand of S1PR1, is a multifunctional lipid mediator and an angiogenic factor. S1PR1 is a dominant S1P receptor in many tissues and an important regulator of lymphocyte trafficking, as well as vascular permeability. Activated S1PR1 induced cortical actin reorganization and increased vascular barrier function.18,27 Despite the abundant expression of S1PR1 and S1PR3 in lung tissue and lung vascular endothelium, only the ligation of S1PR1 reduces murine lung vascular permeability in response to LPS, 17 ischemia/reperfusion,46,47 ventilation-induced lung injury (VILI), 48 and ionizing radiation. 28 Maintenance and formation of adherens junctions was dependent on S1PR1 signaling initiated not only by ligand, but also by fluid shear stress. Changes in the vascular architecture of ECs from S1PR1–/– mice were accompanied by increased vascular permeability, resulting from altered VE cadherin localization at endothelial cell-cell junctions.49,50 Furthermore, S1PR1 ligation is critical to hepatocyte growth factor-mediated, hyaluronan-mediated, and activated protein C-mediated EC barrier enhancement via S1PR1 transactivation.18,19,35,51 In a previous study, our lab had employed a LPS-induced ALI model and studied mice pretreated with simvastatin or placebo prior to the intratracheal administration of LPS. Markers of lung injury including Evans blue albumin dye extravasation, BAL fluid neutrophil and albumin content, and lung tissue histology demonstrated protection from vascular leak and inflammation in mice pretreated with simvastatin. 12 In this study, we further investigated the cell signal pathway-related mechanisms. We demonstrated that simvastatin significantly augments the effects of S1PR1 on the endothelium barrier enhancements, which contributes significantly to the attenuation of vascular permeability. These enhancements were significantly attenuated by KLF2 silencing, which indicated that KLF2 significantly contributed to the effects of simvastatin on gene expression and function of S1PR1 in lung endothelium.
Several studies have suggested that receiving statins improves clinical outcomes 52 of patients with sepsis or ARDS.14,52,53 Pretreatment with simvastatin 24 h significantly reduced LPS-induced lung inflammation in healthy volunteers 15 and patients with severe sepsis-associated ARDS who received statin therapy had a significantly better 28-day survival rate. 54 However, two recent randomized clinical trials showed no improved clinical outcome in patients treated with rosuvastatin 55 or simvastatin 56 within 48 h of onset of ARDS. Therefore, controversy remains regarding the vasculo-protective effect of simvastatin in murine and human inflammatory diseases including ARDS. While there is no satisfactory explanation concerning these contradictory results, the severity of ARDS and timeline of statin treatment may be relevant to these results. The effects of statins on pulmonary arterial hypertension (PAH) are controversial too.57,58 More large and well-conducted clinical trials are needed for further assessing the effects of statins in patients with ARDS and PAH.
In this study, we demonstrated pleiotropic roles of simvastatin in endothelial barrier regulator S1PR1 transcription and expression via KLF2, and their synergic functions on endothelial barrier enhancement. However, there are some limitations in the study. How KLF2 regulate simvastatin induced S1PR1 expression in vivo could be further studied by over-expressing or silencing KLF2 in KLF2 transgenic or conditional knockout mice, the levels of S1PR1 and KLF2 transcription in vivo could be further validated by RT-PCR, and the more detailed roles and mechanisms of simvastatin in regulating S1PR1 by KLF2 in pulmonary and cardiovascular inflammatory diseases need to be further investigated.
In summary, we explored the mechanism of the regulation of S1PR1 expression by simvastatin. We confirmed that simvastatin significantly increased S1PR1 expression by increased KLF2 expression and recruitment of KLF2 to S1PR1 promoter, leading to increased S1PR1 promoter activity. Consequently, pre-incubation of simvastatin significantly increased S1PR1 expression and endothelial response to S1PR1 agonists. These events could significantly contribute the simvastatin and S1PR1 effects on endothelial barrier protection. This study indicated that simvastatin could synergize with S1P to enhance the vascular endothelial function, and be used to reduce the dysfunction of vascular endothelium in multiple inflammatory diseases.
Footnotes
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This work was supported by National Heart, Lung and Blood Institute grants P01HL58064, P01HL98050, and P01HL126609.