
Abstract
Late-onset pulmonary arterial hypertension (PAH) is a rare but fatal complication in patients with childhood surgical repair of dextro-transposition of great arteries (D-TGA), especially with the Mustard and Senning procedures. The pathogenic mechanisms of PAH in patients with repaired D-TGA are not well understood and treatment is not standardized. In this manuscript, we present a case of late-onset PAH in an adult D-TGA patient after Mustard repair and discuss the pathogenic mechanisms, diagnosis, and treatment of pulmonary hypertension in repaired D-TGA.
Introduction
D-transposition of great arteries (D-TGA) is a rare congenital heart condition, with an estimated 1250 babies born in the United States each year affected by this condition. 1 Unlike L-TGA where there is atrioventricular and ventriculoarterial discordance (the ventricles are flipped so the morphologic right ventricle pumps oxygenated blood to the aorta and the morphologic left ventricle pumps de-oxygenated blood to the lungs), in D-TGA there is isolated ventriculoarterial discordance. This means the aorta arises from the right ventricle and the pulmonary artery arises from the left ventricle. The pulmonary and systemic circulations are in parallel and this condition is not compatible with life unless there is associated presence of another shunt to allow mixing of blood.
D-TGA remained a fatal disease with 90% mortality within the first year of life 2 till the introduction of the Mustard and Senning procedures in the late 1950s. In both procedures, a baffle is created within the atria and the deoxygenated blood is redirected from the vena cava to the mitral valve and the oxygenated blood from the pulmonary circulation is redirected to the tricuspid valve. As a result, the anatomic left ventricle pumps the deoxygenated blood to the pulmonary circulation and the anatomic right ventricle pumps the oxygenated blood to the systemic circulation.
The Mustard and Senning procedures were abandoned in favor of the Arterial Switch Operation due to complications associated with the former and better outcomes with the latter. With atrial level repairs, late survival is 84–95% at ten years and 76–89% at 15–20 years, with a steady decline in survival of 0.5% per year.3,4 It is estimated that currently 9000 patients exist in the United States with these atrial level repairs. 5 About 90–95% of adults with atrial level repairs are relatively stable with New York Heart Association (NYHA) functional class I/II symptoms and are able to function as productive members of society. 5 Arrhythmias, late systemic right ventricular dysfunction, baffle obstruction, and baffle leak are well-described, common complications of Mustard repair. 5
Late-onset pulmonary arterial hypertension (PAH) is a rare but serious complication in patients with childhood surgical repair of D-TGA. The pathogenic mechanisms of PAH in patients with repaired D-TGA are not well understood and treatment is not standardized. We present a case of late-onset PAH in an adult with repaired D-TGA and discuss the pathogenic mechanisms of pulmonary hypertension in repaired D-TGA.
Case description
A 46-year-old Caucasian man with a history of repaired D-TGA presented to our clinic with complaints of exertional dyspnea and fatigue. He underwent a Mustard atrial baffle procedure when he was 18 months old. He had an intact ventricular septum, normal pulmonary artery pressure (PAP), and no pulmonic stenosis at the time of the Mustard procedure. The postoperative period was complicated by a stroke with right upper extremity weakness. He had regular follow-ups in a pediatric cardiology clinic until the middle of his second decade, but then was not seen for more than 20 years. Six months prior to this presentation, he noticed increased shortness of breath on moderate exertion and was found to be in NYHA functional class II. Echocardiogram revealed systolic dysfunction of the systemic right ventricle. PAH was not suspected based on sub-pulmonary velocities on echo. He was started on a low dose beta-blocker and angiotensinogen-converting enzyme (ACE) inhibitors, in addition to his aspirin. However, he continued to have progressive increasing exertional shortness of breath. He had no orthopnea or paroxysmal nocturnal dyspnea, chest pain, syncope, or peripheral edema. His family history was significant for D-TGA in his maternal cousin.
On examination, his blood pressure was 110/72 mmHg, heart rate was 68 beats per minute, respiratory rate was 20 per min, and oxygen saturation was 93% on room air. He had no cyanosis, clubbing, jugular venous distension, or lower extremity edema. He had no palpable second heart sound or thrill. Cardiac auscultation revealed normal first heart sound and a loud pulmonic second heart sound with a 3/6-holosystolic murmur on the lower right sternal border. He had normal breath sounds bilaterally. He had residual right upper extremity weakness with no other focal neurological findings. Electrocardiogram showed sinus bradycardia with right axis deviation, incomplete right bundle branch block, and right ventricular hypertrophy with strain pattern. Echocardiogram revealed a hypertrophied systemic right ventricle with moderately diminished systolic function, moderately dilated and hypertrophied pulmonary left ventricle with moderately reduced systolic function, and moderate pulmonic insufficiency. Cardiac magnetic resonance imaging (MRI) confirmed the presence of D-TGA corrected with Mustard atrial baffle procedure (Fig. 1). His systemic right ventricular function was low normal with an ejection fraction (EF) of 52% and his pulmonary left ventricle was moderately dilated with moderately reduced function, with an EF of 42% (Fig. 1). There was no evidence of obstruction in both the systemic venous and the pulmonary venous baffles.
(a) Four-chamber view of the cardiac MRI showing the atrial baffles and moderately dilated sub-pulmonic left ventricle. (b) Short-axis view showing “normal” left ventricle as opposed to a compressed left ventricle, which is seen in patients with D-TGA and atrial switch without pulmonary hypertension. LV, left ventricle (sub-pulmonic); RV, right ventricle (sub-aortic); AB, atrial baffle.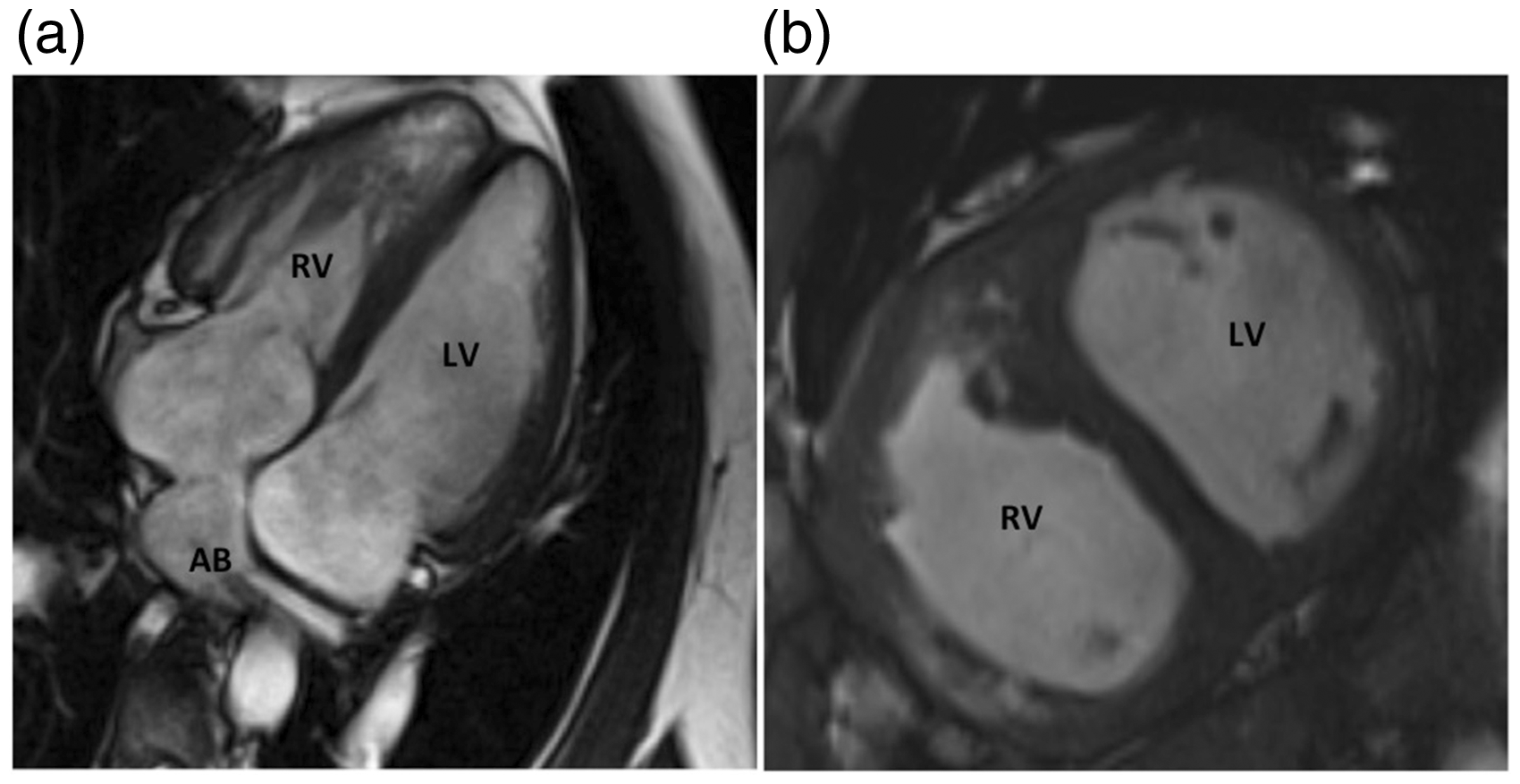
On invasive hemodynamic assessment, his right atrial pressure was 8 mmHg, PAP was 134/49 with a mean of 79 mmHg, and pulmonary capillary wedge pressure (PCWP) was 14 mmHg. Cardiac output as measured by thermodilution was 3.8 L/min with a cardiac index of 1.9 L/min/m2. The pulmonary vascular resistance (PVR) was 17 Wood units. With inhaled nitric oxide, there was no acute vasodilatory response. His O2 saturations were as follows: superior vena cava 72%, right atrium 73%, pulmonary left ventricle 70%, pulmonary artery 67%, and systemic saturation 95%. There was no significant demonstrable left-to-right shunt, right-to-left shunt, or baffle leak (Qp/Qs was 1). His baffle pressure was 12 mmHg. There was moderate superior vena cava baffle stenosis with a 4-mm gradient across the superior vena cava with decompression via azygous veins. He had no inferior vena cava baffle obstruction. There was a mild 6 mmHg gradient across the pulmonary venous baffle to the systemic right ventricle. Invasive angiography confirmed the moderate superior vena cava baffle stenosis but the pulmonary venous baffle could not be well visualized during the levophase. Coronary angiogram revealed normal coronary arteries.
Ventilation perfusion scan excluded chronic thromboembolic pulmonary hypertension. His lung function test showed no evidence of restrictive or obstructive disease. His serology for antinuclear antibody, rheumatoid factor, human immunodeficiency virus, and hepatitis were negative. He had no history of obstructive sleep apnea, but did not have a formal sleep study. Electrolytes, renal function, and liver function were within normal limits. Serum brain natriuretic peptide level was elevated at 548 pg/mL. Normal PCWP excluded pulmonary venous baffle obstruction. He was diagnosed with late-onset PAH associated with repaired D-TGA. He was initiated on upfront combination therapy with Ambrisentan and Tadalafil. Three months after the onset of therapy, his six-minute walk distance improved from 437 m to 458 m but there was no significant improvement in his pulmonic left ventricular ejection fraction by cardiac MRI. On invasive hemodynamic assessment, his PAP decreased to 86/36 with a mean of 53 mmHg, PVR decreased to 9.7 Wood units, and cardiac output increased to 4.8 L/min.
Discussion
Pulmonary hypertension is rare but can be a fatal complication of atrial switch repairs for D-TGA. Multiple mechanisms have been related to the development of pulmonary hypertension in D-TGA, including repair after two years of age, having shunts before repair and elevated pulmonary pressures following surgery. 6 PAH, in the absence of anatomical causes such as pulmonary venous baffle obstruction, has been reported in 7% of patients with D-TGA and Mustard repair. 7 The presence of a left-to-right shunt at ventricular, atrial, or aorta/pulmonary artery level before Mustard repair is associated with increased risk of pulmonary vascular remodeling. 8 The increased pulmonary blood flow resulting from the left-to-right shunting has been suggested to trigger pulmonary vascular remodeling through shear stress-induced endothelial dysfunction. Consistent with this hypothesis, patients with D-TGA who have pulmonic stenosis or who undergo pulmonary artery banding have less advanced pulmonary vascular remodeling (Heath Grade 1 or 2). 8 Children undergoing Mustard repair after the age of two years are more prone to developing pulmonary hypertension as adults, 7 which lends support to the theory that the increased blood flow through pulmonary vasculature causes pathological changes in the vasculature that have reached an irreversible stage by the time the repair is performed. 9 Mild elevation in pulmonary pressures at early postoperative catheterization is predictive of developing pulmonary hypertension as adults. 10
However, increased pulmonary blood flow does not seem to be the only factor leading to increased pulmonary pressures. Children undergoing TGA repairs as early as four days of life can go on to develop pulmonary hypertension. 11 While it is rare, similar to our patient, pulmonary vascular remodeling has also been reported in patients with D-TGA who had an intact septum with no left-to-right shunting. 12 Furthermore, isolated ventricular septal defects with normally oriented arteries rarely induce changes in pulmonary vascular bed despite increased flow within the first year of life. 9 Finally, PAH has also been reported in patients with D-TGA who had undergone arterial switch operation within six weeks of birth. 13 These reports suggest the possibility of other factors in play including genetic predilection or an in-utero condition, predisposing the infant with D-TGA to irreversible pulmonary vascular remodeling. Increased pulmonary blood flow in the fetus and the distribution of oxygenated blood through the fetal pulmonary vascular bed in utero is thought to play a role in the development of PAH later in life. 10
Echocardiography is the screening test of choice for diagnosing pulmonary hypertension in patients with D-TGA. Flattened interventricular septum, dilatation, and dysfunction of the sub-pulmonic left ventricle, as well as elevated sub-pulmonic left ventricular systolic pressure are indicative of PAH. Right heart catheterization is the gold standard for diagnosing PAH in patients with D-TGA as it allows direct measurement of PAPs, PVR, PCWP to identify venous baffle obstruction, and oximetry to exclude left-to-right shunting. In addition, the response to nitric oxide can be used to guide treatment strategies.
The experience with pulmonary vasodilator therapy in patients with late-onset PAH after D-TGA repair is limited to case series. In the study by Cordina et al., 14 of four patients with D-TGA, all were unresponsive to first-line therapy with bosentan, but two patients responded to combination therapy with bosentan and sildenafil. Yehya et al. reported improved functional class with oral pulmonary vasodilator therapy, either mono- or combination therapy, in six patients. 15 Intravenous prostacyclin has also been used to treat patients with repaired D-TGA. 16
Conclusion
The development of PAH after repair of D-TGA is not well understood, but it is associated with a high mortality. Patients after D-TGA repair should be screened for PAH routinely with serial echocardiograms every three years during regular follow-up. As was the case in our patient, echo may miss the diagnosis of pulmonary hypertension, and thus, symptoms of shortness of breath should be evaluated with cardiac catheterization. Early diagnosis and aggressive combination therapy for PAH could help to improve survival in these patients.
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
Funding
TT was funded by an AHA Scientist Development Grant 15SDG25560048 and a Lillehei Heart Institute High Risk and High Reward Grant.