
Abstract
The many types of pulmonary hypertension (PH) are so protean in their biological origin, histological expression, and natural history that it is difficult to create a summary picture of the disease, or to easily compare and contrast characteristics of one type of PH with another. For newcomers to the field, however, such a picture would facilitate a broad understanding of PH. In this paper, we suggest that four characteristics are fundamental to describing the nature of various types of PH, and that taken together they define a number of patterns of PH expression. These characteristics are histopathology, developmental origin, associated clinical conditions, and potential for resolution. The “snapshot” is a way to concisely display the ways that these signal characteristics intersect in select specific types of PH, and is an effort to summarize these patterns in a way that facilitates a “big picture” comprehension of this disease.
Introduction
Summary of the characteristics which constitute the pattern of PH expression.

For each type of PH, four characteristics are used to describe the pattern of PH expression.
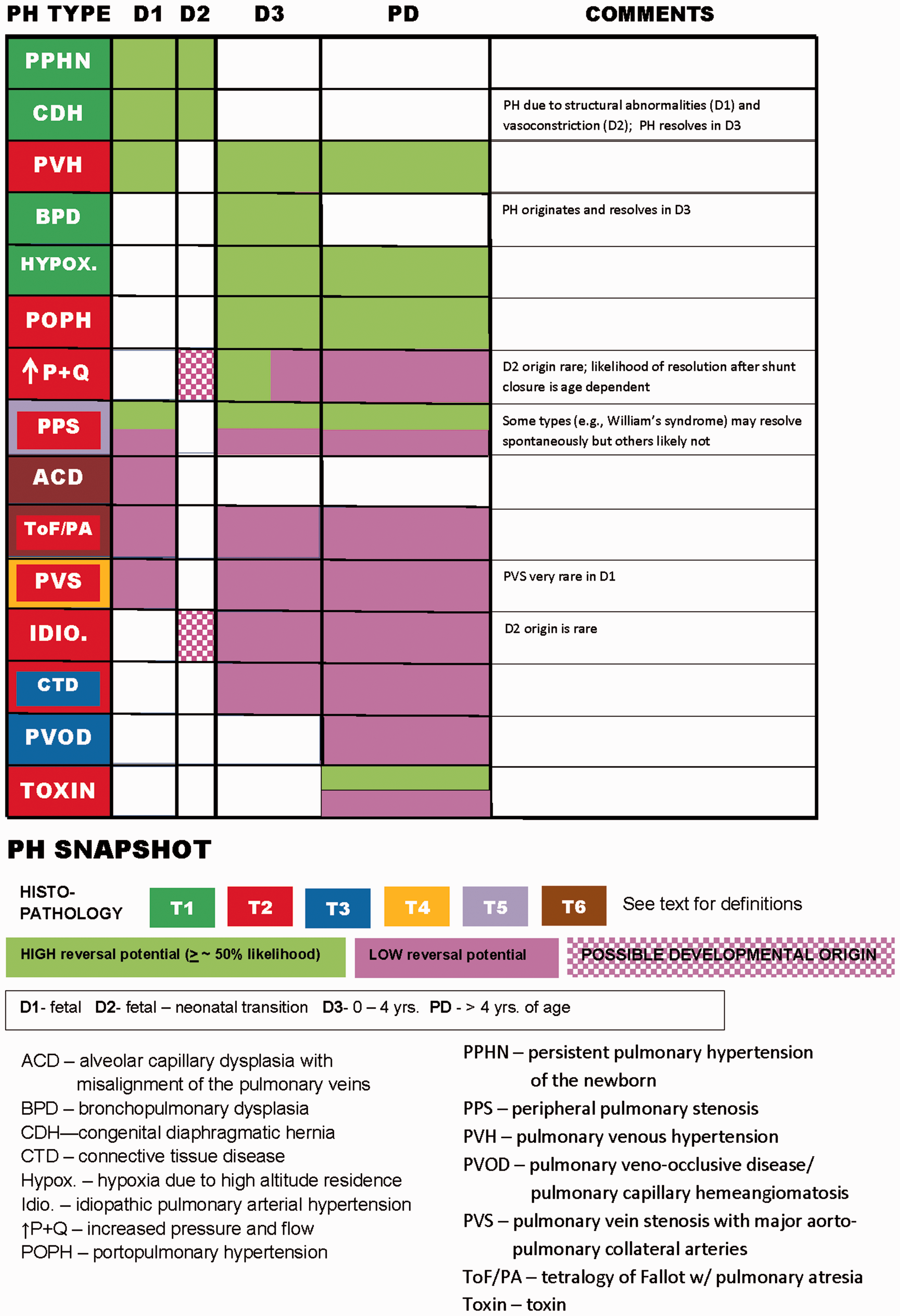
PH snapshot. The background color for the various types of PH (leftward most column) denotes the histopathology, while the color in the columns specifying the developmental period indicates whether the type of PH is potentially reversible.
Definitions and assumptions
Histopathological types
The nature of various types of PH is revealed in part by their microarchitecture, which we divide into histopathological types 1–6. 7 Types T1–3 are derived from the standard approach to classifying pathological changes, where the architecture of small pulmonary arteries (PAs) and pulmonary veins (PV) is emphasized. Admittedly, these descriptions are incomplete, e.g. the number of PAs is seldom included in histological reports, although a reduction in their number is a component of many types of PH,8–10 some structural features (e.g. pulmonary artery–systemic shunt pathways 11 and adventitial thickness 12 ) are not described, and detailed cellular and molecular characterization of the pathology7,13 is too sparsely reported to be included in the snapshot. Other types of microvascular pathology may need to be added in the future as more is learned (e.g. vascular remodeling with cavopulmonary circulations 14 ). Perhaps most important, and as demonstrated in the snapshot, histopathology may correlate poorly with clinical outcome and often does not discriminate between very different types of PH.
T4–6 vasculopathies are relatively rare, encountered mostly in pediatrics, and usually receive little attention in PH reviews but are included because any broad overview of PH ought to include all well-characterized varieties of vascular disease, at least those which occur with some frequency. And these exotic vasculopathies may provide insights regarding more commonly encountered types of PH.
Specific histopathological types are:
T1: distal extension of smooth muscle and medial hypertrophy in small (<100 um) PAs but no intimal or higher grade lesions. 12
T2: increased medial smooth muscle plus intimal hyperplasia and occlusion, ± higher grade lesions (“plexiform arteriopathy”). 12
T3: vasculopathy with intimal occlusion of small PVs playing a major role (pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis [PVOD/PCH, referred to as PVOD here). Exuberant alveolar capillary proliferation also occurs, and T2 lesions in small PAs, although not plexiform lesions.2,12,15
T4: stenosis of large pulmonary veins (pulmonary vein stenosis [PVS]). PVS is mostly a pediatric disease and when observed in adults is generally due to factors iatrogenic or external to the PV.16,17 Leaving aside those forms of this disease, PVS is observed in four settings: following surgical manipulation of anomalously connected PV;18,19 in ex-premature infants, often with bronchopulmonary dysplasia;20,21 with congenital heart lesions without abnormally-connected PV or manipulation of them; and with no obvious associated factors.16,20,22 (A possible link between PVS and other abnormalities, e.g. necrotizing enterocolitis, has been suggested but not proven.)
This vasculopathy has variable expression, ranging from a single discrete area of narrowing of the PV at its junction with the left atrium to diffuse narrowing extending deep into the lung.23–25 The PVs are narrowed/obstructed by intimal and medial myofibroblast proliferation and deposition of carboxylated and sulfated acid mucoploysaccharides.24,26–28 There is also T2 pathological remodeling of small PVs and PAs.22,28 Whether this arterial and small vein remodeling is due to mechanical forces (pulmonary venous hypertension and/or increased pressure and flow [↑P+Q] in unobstructed segments of the lung) or rather reflects a pan-vascular pathology (or both) is unknown. Since anywhere from one to four veins may be affected, this disease is unlike most other pulmonary vasculopathies, where PVs are more uniformly involved.
T5: pulmonary vasculopathy involving narrowing/occlusion of large PAs (peripheral pulmonary stenosis [PPS]). The morphology is variable and can include discrete areas of narrowing in the central PAs, multiple narrowings in lobar and elastic PAs (often but not always at branching points), occlusion of elastic PAs, and diffuse narrowing of lobar and smaller PAs. Post-stenotic areas of PA dilation are also seen. This vasculopathy is observed in a variety of settings, especially Williams syndrome, non-Williams heritable PPS, Alagille’s syndrome, congenital rubella syndrome, moyamoya disease, idiopathic PPS, and with a variety of congenital cardiac malformations. 29
Descriptions of the histopathology of PA stenosis or hypoplasia are somewhat variable. 30 Intimal thickening, fibromyxomatous or cellular,30–34 is the most consistent finding. Also observed is medial hyperplasia,30,35 paucity or fragmentation of medial elastic fibers and mosaic pattern of medial elements,30,31,33–36 and dilation with medial atrophy distal to areas of narrowing.31,33 Hypoplasia of central PAs, with normal intima, media, and adventitia, has also been observed. 30 Since “non-protected” (by large PA stenosis) regions of the vascular bed are often pathologically remodeled (T2 histopathology), presumably due to ↑P+Q, PH is due to both large and small PA pathology.32,37
The variability in macroscopic and microscopic expression is perhaps not surprising, since different gene mutations (with different cognate proteins) are linked with associated conditions—William’s syndrome (ELN 38 ), Alagille’s syndrome (JAG 1 39 ), and moyamoya (RNF213, BRCC3/MTCP1, GUCY1A3 40 ). Indeed, one group found that large PA architecture with PPS subtly differs depending upon the associated condition (Williams, non-Williams, and non-syndromic arteriopathy versus Alagille’s versus moyamoya). 41 For most of the reports cited above, no information is provided regarding any associated diagnoses, so the histological findings cannot be related to a particular variety of PPS, although one report of patients consistent with William’s syndrome described a “mosaic” pattern of elastic tissue in the media. 35
T6: PH associated with disordered vascular architecture due to abnormal development in utero. This type of vasculopathy displays an abnormal spatial arrangement of small PAs and/or veins, although there are features of T1/T2 vasculopathy as well. Two examples are well-described:
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD): malpositioned PV run with the arteries (sometimes in the same adventitial sheath) and bronchi, and there is medial hypertrophy of small PAs with distal extension of smooth muscle, a decreased number of capillaries, thickened alveolar septa with a thickened air-capillary interface, and alveoli and bronchioles are abnormal in shape and number.42–44 Pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries (ToF/PA): pulmonary perfusion is provided in two ways, by central PAs supplied with blood from one or more major aortopulmonary collateral arteries (MAPCAs) ± a patent ductus arteriosus, and directly by MAPCAs, which ramify into small PAs with similar structure (albeit smaller diameter) as normal vessels.
45
The central PAs are smaller in diameter than normal PAs and abnormally distributed. The MAPCAs are prone to develop intimal thickening and occlusive cushions anywhere along their course, limiting or interrupting perfusion to the segment(s) of the lung so supplied. And when MAPCAs do not narrow, T1/T2 vascular disease develops in the distal vessels due to ↑P+Q.45–48
Developmental origin
Developmental events clearly underlie a few types of PH (e.g. ACD), but their significance is likely more general; appreciating how development goes awry may also inform our understanding of PH originating in the fully-developed circulation 7 and suggest avenues for reconstructing the pathologically remodeled vascular bed. For the snapshot, PH can originate during one or more of four phases: fetal (D1); transitional (D2); developmental (D3); or post-development (PD). Fetal development confers abnormal pulmonary vascular structure of fetal origin. In utero events, which increase the risk of post-natal PH but are not necessary for it to develop (e.g. chorioamnionitis),49–51 are not considered, nor is developmental programming, as the latter is too little understood at this time to permit inclusion. The fetal–neonatal transition—the rapid and radical in reduction of pulmonary vascular resistance (PVR) immediately after birth—is a critical physiological/structural developmental milestone.52–55 The developmental phase is difficult to define precisely as lung development starts in utero but continues well after birth. 56 No sharp line separates the developing from the developed lung. Alveoli and supernumerary arteries increase in number until approximately eight years of age, but the most rapid vascular growth occurs earlier: alveoli and conventional PAs multiply most rapidly in the first 18 post-natal months, while for intra-acinar arteries rapid growth continues until about four years of age.56,57 For the snapshot, the first four years are therefore considered the developmental phase, recognizing that growth continues beyond that time (post-developmental phase, for purposes of the snapshot).
Several types of PH develop in the first few years of life, but it is unclear exactly how the fact of development per se influences the pulmonary vasculopathy in these situations. For example, could incomplete resolution of fetal PVR be the origin of idiopathic PH in a four-month-old? Do ↑P+Q cardiac shunting defects cause more (or less) severe alteration of the pulmonary circulation because they impact the developing lung? Studies finding fewer small PAs in patients with cardiac shunts, not explained by intimal proliferation, are consistent with the notion that ↑P+Q impedes small PA development,9,58,59 but since essentially all our experience with this hemodynamic alteration originates in the developing organism, it is hard to confirm this. Also, with some types of disordered development (e.g. congenital diaphragmatic hernia [CDH] and bronchopulmonary dysplasia [BPD]) PH often resolves in the first few years, indicating that impaired early development is not always permanent. Indeed, it seems likely that resolution of some types of PH (e.g. CDH) is actually facilitated by ongoing development.
Associated clinical conditions
A major preoccupation for the field has been arranging into categories PH associated with other medical conditions—indeed, that is the main feature of the Nice classification. Since, with few exceptions (e.g. ↑P+Q), PH occurs in only a minority (usually very small) of individuals with a PH-associated illness, be it other organ system disease, toxins, infections, environmental factors, or gene mutation, these conditions are actually risk-modifying, not causative factors,2,60 which require interaction with other elements to culminate in PH. The clinical importance of this taxonomy is obvious, but associated conditions are incorporated into the snapshot for a second reason as well: connecting PH with associated illnesses has, and will, help us better understand the pathobiology of PH. An obvious example is pathological remodeling due to ↑P+Q (almost always due to congenital cardiac malformations). 61 Though its mechanism(s) is unclear, it is certain that these mechanical factors are powerful stimuli for remodeling, a fact which was revealed when early investigators considered the effects of congenital cardiac defects on the lung’s circulation. And its relevance extends well beyond cardiac disease since these mechanical factors presumably operate with any type of pulmonary vasculopathy with uneven distribution of Q and P, as noted below.
The approach used here is similar to that used in the Nice classification insofar as select disease entities associated with PH (e.g. portopulmonary hypertension [POPH]) are incorporated, but it differs in one respect. Physiological perturbations (↑P+Q, pulmonary venous hypertension, alveolar hypoxia) are included rather than their cognate clinical conditions (e.g. congenital septal defects, left heart disease, lung disease), because specifying the responsible biological abnormality identifies the presumed proximate cause of the PH.
Potential for resolution
Types of PH for which PA pressure falls to normal, or nearly so, spontaneously, after a brief period of therapy (e.g. persistent pulmonary hypertension of the newborn [PPHN]) or after the inciting stimulus is removed (e.g. surgical relief of pulmonary venous hypertension via mitral valve intervention), are considered to have high potential for resolution. Since nearly any type of PH can rarely and unpredictably resolve, 62 the likelihood of this needs to be specified, and we regard ≥ ∼50% odds of resolution as sufficient to qualify for high potential. “Resolution” does not necessarily imply completely normal PAP or PVR. While there are ample hemodynamic data on most types of PH upon first diagnosis, follow-up catheter-derived information is limited for most varieties, and since available echocardiographic data do not specify the exact PAP, mildly elevated PAP may escape detection. Nor does it imply that the pulmonary circulation is functionally normal. Even if normal at rest, PAP may increase abnormally with exercise 63 and/or the lung may be hyper-reactive to vasoconstrictive stimuli. Nor does it necessarily indicate complete histological normalization. For example, with mitral stenosis, even patients with biopsy-proven intimal fibrosis before relief of the stenosis show marked reduction in PAP after repair 64 and the same has been described with intracardiac shunts operated early on, 9 but it seems unlikely that the intimal lesions disappear completely. This definition therefore lacks biological rigor and could be misleading insofar as it might seem to imply a completely normal clinical phenotype. But it does distinguish progressive PH from more benign varieties, including the potential for a pulmonary circulation compatible with treatment-free, symptom-free existence.
What the snapshot does not do
It does not include all types of PH, rather only those varieties—somewhat arbitrarily chosen—which represent particular intersections of properties and for which there is sufficient information to adequately characterize the PH. The snapshot is not a comprehensive classification scheme, nor an alternative to or replacement for existing taxonomies.2,5 That said, the distinct combinations of snapshot characteristics could be affixed to any type of PH and it so characterized, e.g. idiopathic PH would be T2, D2–PD, irreversible; POPH would be T2, D3–PD, portopulmonary, reversible. It does not incorporate notions of “treatability”. (i) Therapeutic approaches are rapidly evolving and FDA-approved targeted PH therapy has oozed across Nice categories (e.g. riociguat for class IV PH). And even where consensus indications for targeted therapy are lacking (e.g. Nice groups 2 and 3), this may attest to a lack of trials utilizing suitable patients rather than the lack of biological effect.2,65 (ii) It is usually unclear what the clinical effectiveness of a drug (generally measured with functional criteria only partly influenced by the state of the pulmonary vascular bed) tells us about the nature of the PH being treated. Targeted PH therapy may have salutary effects on organ function other than the lung (e.g. the effect of PDE5 inhibitors on ventricular function) which account for a significant fraction of their benefit. The snapshot does not take into account acute reactivity to vasodilators. This is an important characteristic for individual patients since it separates patients within a type (especially idiopathic pulmonary arterial hypertension) with respect to response to therapy and survival.66,67 But it is of little utility in distinguishing between different types of PH since it is observed in some patients with many different types of PH. Indeed, it is possible that reactivity is usually present in the earliest stages in most or all forms of PH. And reactivity is usually lost,66,67 which complicates trying to incorporate it into a summary classification. Transient PH is not incorporated into the snapshot since in most or all types of PH (and the healthy pulmonary circulation, for that matter), PVR can acutely and reversibly increase with provocation (e.g. alveolar hypoxia). The snapshot will not have long-term value without modification and enhancement. As more is learned of the developmental origins of pulmonary vascular disease, as better ways of defining its histopathology are developed, and as detailed mechanistic pictures of the various types of PH are uncovered this information will need to be incorporated into updated snapshots.
What the snapshot does do
It summarizes how many of the various types of PH are similar, and differ, with respect to the four important characteristics addressed here. It therefore provides a fairly broad but concise overview of this arena, to our knowledge one not previously available. The field is currently focused mostly on idiopathic and other types of persistent PH, but the snapshot emphasizes the fact that many types of PH are reversible, at least if inciting causes can be eliminated, including those associated with T2 histopathology. The potential for favorable remodeling (even if this cannot be effected as often as we would like) is thus emphasized and gives reason for optimism for eventual development of definitive therapies. The snapshot draws attention to forms of vasculopathy little discussed in major reviews (e.g. T4 and T5) and emphasizes how dynamic the pulmonary vascular bed can be (where developmental changes can greatly alter the characteristics of the lesser circulation). We hope that this stimulates new ways of thinking about the biology of pulmonary vascular remodeling and perhaps even new ideas for research.
Types of PH: rationale for assignment of characteristics
Idiopathic persistent pulmonary hypertension of the newborn (PPHN)
There are many conditions associated with severe PH in the neonate, but this summary focuses on the idiopathic variety, where the chest X-ray is normal (although there are often associated perinatal complications).52,68
Histopathology: T1. Distal extension of smooth muscle into normally non-muscular arteries, increased thickness of small arteries, and increased adventitial thickness are the typical findings.69–71 Two case reports also described loss of small PAs, one with idiopathic PPHN 71 and another with PPHN clearly associated with premature ductal restriction. 72
Developmental origin: D1–2 (origin and resolution). It is unclear what fraction of idiopathic PPHN has fetal origins as distinct from being purely a reflection of a disordered transition. The authors of three histological studies suggested that the magnitude of wall thickening and distal extension in small PAs suggest that the disease originated in utero.69,70,73 But the number of histological observations are relatively few and the patients, who died, may not reflect the disease in the vast majority who survive. It is also possible that the distal extension of smooth muscle occurred post-natally. 71 More to the point, since the high PVR usually substantially resolves within a few days, it is hard not to think of PPHN as being due to delayed transition, even if set up in utero. 52 The one clear exception is PPHN due to premature constriction of the ductus arteriosus, 74 and therefore fetal origin is also assigned.
Potential for resolution: High. We are aware of no data regarding early (neonatal) survival or PH over the long term for idiopathic PPHN per se in the modern (ECMO and iNO) era, but since early survival of PPHN patients as a class (excepting CDH) is ∼90%75,76 and very few patients have echocardiographically detectable PH on longer-term follow-up,77,78 it is clear that PH usually (if not invariably 79 ) resolves in these patients.
Congenital diaphragmatic hernia (CDH)
Histopathology: T1. Post-mortem examinations have shown, in addition to bilateral lung hypoplasia, a reduction in the number of airway generations and alveoli, reduction in the size and number of small PAs, with increased wall thickness and extension of muscle into smaller arteries than normal.80–82
Developmental origin: D1–3 (resolution in D3). The vascular architectural abnormalities are clearly of fetal origin, but marked pulmonary vasoconstriction, which may resolve, in the early neonatal period83,84 indicates that an abnormal transition can also play an important role.
Potential for resolution: High. While persistent and occasionally severe PH has been described a few years after CDH repair,85,86 the vast majority of survivors (including those requiring extracorporeal membrane oxygenator support adjacent to repair) have non-detectable or only mild elevation in RV pressure.87–92 (The fraction of non-survivors having refractory disease prior to demise is surely much greater, but given an overall survival rate of ∼75%, 87 only a relatively small percent of the population of patients with CDH develops suffers from refractory PH. 91 ) It is possible that mildly increased PAP is under-diagnosed in these patients, but the many outcome reports making little or no mention of PH (e.g. Ssemakula, 93 Davis, 94 Vanano, 95 Muratore, 96 Bagolan 97 ) suggest that clinically appreciable PH in this setting is very uncommon.
Pulmonary venous hypertension (PVH)
Histopathology: T2. Arterial medial hypertrophy, longitudinal smooth muscle bundles, and intimal thickening and fibrosis, sometimes so severe as to obstruct the lumen of small PAs, have been described even in children, but higher grade lesions are not observed.13,64,98 Pulmonary veins are affected as well, with the media (hypertrophy, fibrosis, “arterialization”) and intima (fibrosis and longitudinal smooth muscle cells) both involved.85,99
Developmental origin: D1, D3, PD. Thickening of pulmonary veins with congenital lesions causing pulmonary venous obstruction has been observed at a few days of age;100–102 hypoplasia of small PAs is also described.103,104 Vascular changes also develop in the developing and mature lung. 105
Potential for resolution: High. The great majority of patients who develop PH due to PVH outside the neonatal period have substantial or complete resolution of PH with relief of PVH. 61 There are few data regarding patients with PVH since birth, however, and it is possible that the potential for resolution is less favorable if persisting for years. 106
Bronchopulmonary dysplasia (BPD)
Histopathology: T1. Available histological information largely concerns patients with the “old” BPD, so it is possible that the micro-architecture of “new” BPD may differ. Not all patients evince arterial changes, but those who do show distal extension of smooth muscle, medial hypertrophy, and increased adventitial thickness.107–112 Swollen or “chunky” endothelial cells, which encroach on the PA lumen, are also described,107,110,111 but endothelial proliferation is seldom observed (<12%109,111,112). T1 is therefore assigned as the pathological type, recognizing that a few patients may develop higher grade vasculopathy. Abnormal alveolar capillaries are also described,113,114 but whether this contributes to increased PVR is unclear, especially when one considers that these patients can manifest both “reactivity” to vasodilators 115 and a tendency to markedly increase PAP with viral and other insults. A reduced number of small arteries 8 likely also contributes to increased PVR.
Developmental origin: D3 (origin and resolution).8,116
Potential for resolution: High. The precise likelihood of resolution of PH in these patients is unknown, but published reports indicate that it is very likely greater than 50%.21,117–119
Chronic alveolar hypoxia due to residence at high altitude
Alveolar hypoxia is a component of multiple disorders, e.g. chronic lung disease, but in these cases PH may be partly due to stimuli other than hypoxia per se (e.g. inflammation). We therefore focus on relatively “pure” alveolar hypoxia—that which attends residence at high altitude.
Histopathology: T1. Distal extension of smooth muscle, longitudinal muscle bundles, and (generally mild) medial hypertrophy are typically described.120–122
Developmental origin: D3, PD.123–126
Potential for resolution: High. Relocation to low altitude results in near or complete resolution of PH in the great majority.127–131
Portopulmonary hypertension (POPH)
Developmental origin: D3, PD. PH due to portosystemic venous shunts is rare but well-described in infants and children.134–137 But POPH is far more often diagnosed in adults, without apparent origin in early pulmonary vascular development.
Potential for resolution: High. How often is POPH reversed when its putative cause (portal hypertension) is eliminated by a liver transplant (LT)? Only case reports and small series are available, often with scanty hemodynamic follow-up. Some reports suggest relatively few patients show resolution of POPH after LT.138–140 But three groups with extensive experience reported that targeted PH medications were discontinued in at least 50% of their patients with pre-LT POPH 141–143 (although neither PAP or PVR post-LT were documented for most of the patients). Other small series and case reports show a substantial resolution of POPH, including documentation of RV systolic pressure ± PVR,144–146 but with too few patients to determine the probability of resolution. Michael Krowka estimated that in ∼50% of patients, pre-LT prostacyclin can be discontinued with normal pulmonary hemodynamics after transplant. 147 The precise mechanism(s) driving improvement in pulmonary hemodynamics after LT remain elusive, although immunosuppressive regimens and the reduction of circulating factors derived from the removed diseased liver may both contribute. Intriguingly, the immunosuppressive medication tacrolimus is under investigation as a therapeutic for PAH, which while it may relate to an immune modifying role is being investigated currently more so due to its potential ability to increase molecular BMPR2 expression in PAH cases. 148 It should be appreciated that many cases of POPH resolution involve only mildly increased PVR,142–144,146,149,150 far lower than in other forms of reversible PH; a reduction in PVR from 3 or 4 WU to 2 WU does not imply much of a change in the architecture of the vascular bed. But the fact that POPH patients, prior to LT, often show a marked response to relatively low dose epoprostanol140,149,151,152 suggests that there is less fixed reduction of the vascular bed with POPH than with some other varieties of PH.
↑P+Q
We make no effort to grapple with congenital cardiac defects as a class, as PH-associated conditions, because besides altered P+Q, there are other confusing (e.g. PH with small septal defects) and perplexing factors involved (e.g. propensity for early vascular disease with d-TGA, not explained by hemodynamics153–155). We focus instead on the physiologic abnormality of simultaneously ↑P+Q, with large (pressure unrestrictive) ventricular septal defect (VSD) and patent ductus arteriosus (PDA), being the archetypical types of these malformations. 61 Indeed, there are very few clinical situations where these two hydrodynamic abnormalities occur in concert except with congenital cardiac malformations. Exactly how high is high, with respect to P and Q provoking vascular disease, is unclear but presumably varies somewhat from patient to patient. Most patients who develop vascular obstructive disease with a VSD have a pulmonary to systemic flow ratio > 2 prior to developing the vascular changes, and two reports suggest that fixed vascular changes are very unusual, at least in childhood, with mean PAP < 50 mmHg, 61 although it should not be assumed that PAP near that value is reliably “safe.”
We refer specifically to greatly ↑P+Q, not to increased Q with low PAP; pre-tricuspid left-to-right shunts, which typically initially increase Q without much increasing P, can cause PH, but its likelihood and rate of development are much less than for ↑P+Q suffered from birth. 61 We focus on ↑P+Q because it is a well-defined stimulus, the major factor driving development of vascular disease in many cardiac malformations, and because of its broader significance. This stimulus likely plays a role in many—maybe all—types of PH: in T2 PH there are highly narrowed or obstructed PAs as well as unobstructed vessels;10,12 ↑P+Q in the unobstructed vessels likely leads to secondary pathological remodeling in these previously unaffected arteries.
Histopathology: T2. Patients with ↑P+Q initially develop T1 histopathology, but with time have a high likelihood of developing plexiform arteriopathy.9,12,156,157
Developmental origin: D2–3, PD. Paul Wood thought that patients with Eisenmenger’s physiology had never experienced the normal early post-natal decline in PVR, 158 but it is clear that initially low PVR can increase with time, as does the risk of fixed/progressive vascular disease.159,160 The snapshot acknowledges that high PVR may occasionally have its origin in the transition, but that irreversible remodeling is far more likely to develop in the developmental period or later. (PH caused by ↑P+Q in utero due to premature constriction of the ductus arteriosus is classified under PPHN.)
Potential for resolution: High/low. With a large VSD or PDA even greatly increased PVR can be much reduced or even normalize after repair of the defect if it is closed in the first year or two of life, but in other cases PVR will remain high or increase after repair, especially if closed later.161–168 The snapshot confers this by indicating early potential for resolution which subsequently declines.
Peripheral pulmonary artery stenosis (PPS)
As noted above, PPS is associated with a number of systemic conditions, and different types of PPS may differ significantly in important characteristics. PPS with William’s syndrome is probably the best characterized in terms of hemodynamics and natural history.
Histopathology: T5, T2.
Developmental origin: D1, D3, PD. PPS associated with William’s syndrome has been documented in patients aged < 5 weeks,169,170 and PPS of undefined association shortly after birth36,171 indicating likely in utero development, but progression can occur later.41,171,172
Potential for resolution: High for William’s syndrome but unclear for other types of PPS. Multiple reports suggest that substantial reduction of PH in infancy and childhood occurs in a majority of patients with Williams-related PPS although complete resolution is unusual and progression occurs in some cases.38,41,171–175 The potential for resolution for PPS associated with other conditions is likely much less, but longitudinal information is sparse. The snapshot indicates this by using both pink and light green to indicate the potential for resolution.
Alveolar capillary dysplasia with misalignment of the pulmonary veins (ACD)
Histopathology: T6.
Developmental origin: D1. 43
Potential for resolution: Low. It is widely agreed that this disorder is essentially never reversible,43,176 although it appears that there may be rare variants which do not result in early death.177,178
Tetralogy of Fallot with pulmonary atresia and MAPCAs
Histopathology: T6.
Developmental origin: D1, D3, PD. The architecture of the central PAs/MAPCAs develops in utero, but T1/T2 disease can occur in the early developmental period and later, as noted above. 45
Potential for resolution: Low. In the unoperated patient, this type of pulmonary vasculopathy eventually causes reduced and maldistributed pulmonary blood flow due to narrowing of MAPCAs and vascular disease in ↑P+Q segments.
Pulmonary vein stenosis (PVS)
Histopathology: T4, T2.
Developmental origin: D1, D3, PD. PVS has been described in newborns,23–25 indicating this disease can develop in utero. But it is unknown whether hypoplasia of the veins versus intimal hyperplasia (or both) cause PV narrowing in utero; we know of only one report of histology in a (one-day-old) neonate, 24 which implied that focal PV narrowing at the left atrial junction was due to fibrous intimal thickening. But intimal and medial changes have been described in patients as young as two months,26,27 indicating that intimal hyperplasia surely can occur at least that early. It is conceivable that all PVS originates in the fetus and then progresses, but this seems unlikely: (1) Breinholt 179 described ten patients with no evidence of PVS (by echocardiography) who subsequently manifested the disease; (2) there are multiple reports of PVS presenting after the age of three years;24,179–181 and (3) PVS developing after surgical correction of anomalously connected PV18,19 is certainly a post-natal event. Prenatal and early developmental phase is therefore assigned, with most PVS developing after birth.
Potential for resolution: Low. Patients with one or two stenotic veins may be clinically stable indefinitely, presumably because only a tolerable fraction of the pulmonary circulation is affected, but the authors know of no reports of spontaneous resolution of the disease. When three or more veins are involved, progression of stenosis is almost always relentless.16,23,179,180
Idiopathic pulmonary arterial hypertension (including heritable forms)
Histopathology: T2. 12
Developmental origin: D2 (possible), D3, PD.182,183 There are a few reports of infants with signs/symptoms of PH in the early neonatal period in whom unremitting and ultimately fatal PH is diagnosed weeks or months later,184,185 and patients with apparently idiopathic PH as young as one month of age are included in one study. 182 Since these and a few other reports suggest that incomplete fall of fetal PVR may account for a few cases of idiopathic PH, possible D2 origin is indicated.
Connective tissue disease (CTD) associated PH
Histopathology: T2/T3.12,187,188 In addition to T2 pathological changes, a substantial fraction of patients with CTD of different types have PVOD-like small PV involvement.
Developmental origin: D3, PD.188–190
Potential for resolution: Low. 191
Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis spectrum disease (PVOD)
Histopathology: T3.
Developmental origin: PD.192,193 So few cases of PVOD presenting in the developmental period have been described193,194 (save those secondary to medical therapy) that it seems appropriate to assign post-developmental origin of the disease.
Toxin-associated PH
Histopathology: T2 (aminorex and fenfluramine195,196).
Developmental origin: D3, PD. Many drugs have been reported as possible or definite risk factors for PH, with appetite suppressants being among the best characterized examples of the latter.2,15 Aside from chemotherapy for malignancy in children, 197 the best-described cases of toxin-associated PH have occurred in adults, although diazoxide has been associated with PH in infants.198–200
Potential for resolution: Low/high. Resolution of PH is very unlikely with aminorex and fenfluramine.196,201 On the other hand, diazoxide-associated PH in infants appears to be reversible, although available information is very limited.198–200 Dasatinib-related PH usually substantially improves (or completely abates) following cessation of the drug, although the best available information suggests that PAP usually remains elevated, at least for a few months. 202
Summary
When various types of PH are thought of as having distinctive patterns of expression, and these patterns are graphically displayed, several PH features perhaps not widely appreciated become readily apparent:
While the field is currently focused heavily on idiopathic pulmonary arterial hypertension and other types of persistent PH (e.g. CTD, and CLD), many types of PH are largely reversible if inciting causes can be eliminated, indicating that the pulmonary circulation’s capacity for favorable remodeling is perhaps greater than often perceived. Abnormal pulmonary blood vessels can take many histopathological forms, and with only a few exceptions (pulmonary venous hypertension being the most striking) reversible PH is associated with histopathology largely limited to increased medial smooth muscle. Pulmonary vascular pathology can originate at any time of life, from early fetal life to late in adulthood. The potential for PH resolution is not tightly linked to any developmental period—early, and late, developmental origins are associated with types of PH which do, and do not, resolve.
Footnotes
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.