
Abstract
Abemaciclib (ABE) in combination with endocrine therapy represents the mainstay treatment for either endocrine-resistant metastatic or high-risk early-stage HR+/HER2− breast cancer patients. Hence, an adequate knowledge of this agent pharmacodynamic, pharmacokinetic, and of its drug–drug interactions (DDIs) is crucial for an optimal patients management. Additionally, ABE interference with food and complementary/alternative medicines should be taken into account in the clinical practice. Several online tools allow to freely check DDIs and can be easily consulted before prescribing ABE. According to one of this instruments, ABE display the lowest number of interactions among the available cyclin-dependent kinase 4/6 inhibitors. Still, clinicians should be aware that online tools cannot replace the technical datasheet of the drug as well as a comprehensive clinical assessment for each patient. Here we critically review the main pharmacological features of ABE, then focusing on its potential interactions with drugs, food, and alternative medicine, in order to provide a guide for its optimal use in the treatment of HR+/HER2− breast cancer patients.
Plain language summary
Why was the review done? Abemaciclib, paired with hormone therapy, is a key treatment for breast cancer patients whose cancer cells respond to hormones but not to a protein called HER2. Understanding how this medication functions in the body, how it interacts with other drugs, and how the body processes it is crucial for providing optimal care.
What did the authors do? The authors looked for published evidence about the way abemaciclib works into the body and about how it interacts with other drugs (including alternative medicines) or food. Then they summarized these findings.
What did the authors find? Abemaciclib absorption, distribution, metabolism and excretion is well known and it is here described. What people eat and any alternative medications they take can affect how abemaciclib works. Online tools are available for doctors to check potential interactions between abemaciclib and other drugs a patient might be using. It’s advisable for doctors to consult abemaciclib data sheet and use online tools before prescribing the drug. Notably, compared to similar treatments, abemaciclib has fewer interactions with other drugs.
What does the review mean? This review delves into how abemaciclib works in the body and explore its potential interactions with other drugs, food, and alternative medicines. This information will aid doctors in using abemaciclib effectively for treating breast cancer patients.
Keywords
Introduction
The combination of a cyclin-dependent kinase 4/6 inhibitor (CDK4/6i) with endocrine therapy (ET) represents the current standard of care for hormone receptor positive (HR+), human epidermal growth factor receptor 2 negative (HER2−) metastatic breast cancer (mBC) patients.1,2 Indeed, these regimens improved the survival outcomes of HR+ mBC patients compared to ET alone. 3 However, the three commercially available CDK4/6i [i.e. palbociclib (PAL), ribociclib (RIB), and abemaciclib (ABE)] display a wide inter-individual variability in term of adverse events and tumor responses, due to multiple biological and clinical reasons. 4 Among these factors, pharmacological features and drug–drug interactions (DDIs) may play a significant role, as they may influence CDK4/6i activity and metabolism, potentially increasing toxicity or decreasing efficacy.5–7 In the clinical practice, DDIs are frequent, and their risk is higher in case of polypharmacy (i.e. the concomitant use of five or more drugs), a common condition among elderly cancer patients with several comorbidities.8–10 DDIs can lead to adverse drug reactions (ADRs), which may, in turn, induce further inappropriate prescriptions, a phenomenon known as the ‘prescribing cascade’. 11 According to a recent report, the estimated prevalence of polypharmacy and potentially inappropriate medications in older adults with newly diagnosed cancer are 80% and 41%, respectively. 12 Moreover, in the real-world scenario, ADRs may also be caused by interactions with self-prescribed over-the-counter agents. This is the case of hypertension due to oral decongestants or rhabdomyolysis following statins. Lastly, food and beverages may also determine clinically relevant interactions with anticancer drugs. For instance, grapefruit and pomegranate juice interfere with the metabolism of drugs metabolized by cytochrome P450 3A4 (CYP3A4) enzymes. 13
Given the frequency and potential consequences of the abovementioned interactions, they should always be tested before prescribing a CDK4/6i.5,14 Medical oncologists can prevent significant DDIs and minimize drug toxicities using freely available online interaction checkers.5,15
Among CDK4/6i, ABE is the most potent and represents the treatment of choice for either endocrine-resistant metastatic and early BC patients at high risk of relapse after surgery.16–18 Given the large number of BC patients exposed to ABE, a proper knowledge and understanding of its pharmacological proprieties and of potential interactions with drugs, food, beverages, and complementary agents is of paramount importance in the clinical practice.
In this review, we provide a description of ABE pharmacology, followed by a general overview about DDIs, to then put in context and summarize the main pharmacological interactions of ABE.
ABE pharmacology at a glance
ABE is an orally administered agent, commercially distributed as tablets. The approved starting dose of ABE in combination with ET is 150 mg bis in die (BID) continuously. Two dose reductions, to 100 and 50 mg BID, are allowed in case of toxicities. 19 The main pharmacological characteristics of ABE are described in the following paragraphs.
Pharmacodynamic
By phosphorylating and inhibiting the retinoblastoma (Rb) oncosuppressor protein, CDK4/6 promote cell cycle progression from G1 to S phase, eventually stimulating cell proliferation. 20 ABE exerts its antineoplastic activity through an ATP-competitive reversible inhibition of CDK4 and 6, with an inhibitory concentration 50 (IC50) of 2 and 10 nM, respectively. 21 Among the available CDK4/6i, ABE is the most potent.
Multi-omics studies showed that, at clinically compatible concentrations, ABE displays multiple secondary targets, including CDK1-cyclin B and CDK2-cyclin A/E complexes. This observation explains why ABE can also induce G2 cell-cycle arrest along with a pan-CDK transcriptional signature. 17 CDK9 is also a secondary targeted of ABE. This kinase plays an essential role in the proliferation of intestinal cells, as demonstrated in experimental models.22,23 Additionally, ABE inhibits glycogen synthase kinase-3 beta (GSK3β), which is part of a protein complex that phosphorylates β-catenin, preventing its translocation into the nucleus. GSK3β also complexes with other transcription factors to form transcriptional activators of multiple genes, including MYC, CCND1, and AXIN2. Inhibition of GSK3β induces Wnt pathway/β-catenin activation, favoring cellular proliferation. 24 Moreover, ABE profoundly inhibits Ca2+/calmodulin-dependent protein kinase, which is involved in intestinal motility and linked to bowel movements.22,23 Figure 1 displays ABE pharmacodynamic features.

Pharmacodynamic of abemaciclib. Abemaciclib presents several targets beside CDK4/6. These include – but are not limited to – CDK1-cyclin B and CDK2-cyclin A complexes, CDK9, GSK3β, and CAMKII.
Pharmacokinetics
ABE is highly soluble at a wide pH range (1–6.8) and display a moderate permeability. 25 After absorption from the gastrointestinal tract, ABE enters the bloodstream through the portal system (Figure 2). Average time to maximum plasma concentration is 4 h (0–10.2). 16
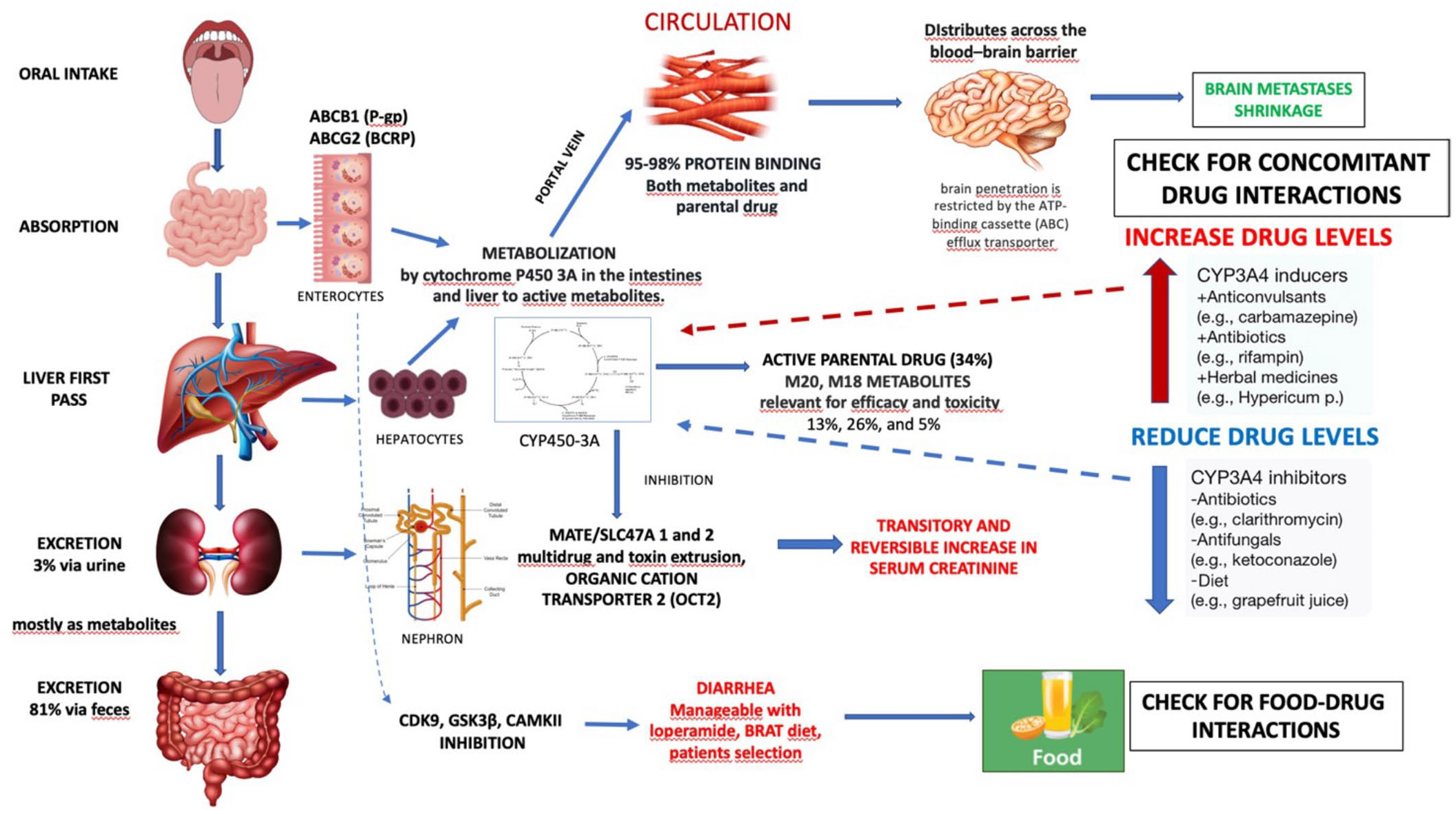
Pharmacokinetic of abemaciclib.
In the systemic circulation, the observed average fraction of protein-bound ABE is 96.3%, mainly to albumin and α-1-acid-glycoprotein, with similar rates for its metabolites. 6 Hence, protein binding influences tissue distribution of ABE.
Additionally, ABE crosses the blood–brain barrier (BBB) by passive diffusion, reaching cerebrospinal fluid concentrations similar to those observed in plasma.16,26 However, ABE diffusion into the central nervous system may be limited by the presence of transmembrane ATP-binding cassette (ABC) transporters, such as P-glycoprotein (P-gp, ABCB1) and breast cancer resistance protein (BCRP, ABCG2), which use ATP hydrolysis to extrude endogenous and exogenous compounds. 27
In vitro and in vivo drug disposition studies demonstrated that ABE is extensively metabolized in the liver via cytochrome P450 CYP3A4, but not CYP3A5. ABE metabolites include N-desethylabemaciclib (M2), hydroxyabemaciclib (M20), and hydroxy-N-desethylabemaciclib (M18). 28 These oxidative derivatives are present in significant concentrations in plasma and account for approximately 45% of total plasma radioactivity, according to human mass balance studies. Moreover, these metabolites have a higher affinity for CDK4 and CDK6 compared to ABE, with lower IC50. 28
Excretion of ABE and its metabolites mainly occurs through the biliary tract (97%), whereas renal elimination is negligible (3%). Several transporters also play a role in ABE excretion, as they are expressed in hepatocytes and nephrons, where they extrude the drug and its metabolite from blood into the urine and bile, respectively. 29 After a single-dose, ABE half-life is 22.8 h (range 8.9–60.8). 16
Overview of pharmacological interaction mechanisms
In general, DDIs are caused by pharmacodynamic or pharmacokinetic interactions that may alter plasmatic drug levels. This may increase usually mild or negligible side effects or, on the contrary, decrease a drug activity. 30 Here, we discuss the main features of pharmacological interactions, providing relevant examples relative to CDK4/6i, when appropriate.
Pharmacodynamic interactions
Pharmacodynamic interaction occurs when two agents are given together and exert their action at the same or similar receptor site, leading to an additive/synergistic effect or to an antagonist effect. Hence, evaluation of pharmacodynamic DDIs in the clinical practice may reduce the risk of toxicities and improve the quality of patients’ care. 31
A paradigmatic example of pharmacodynamic-based DDIs occurring in BC patients is the potential cardiac toxicity stemming from the concomitant administration of RIB and QT-prolonging drugs. 32 Recent reports confirm that ABE is devoid of such toxicity. Indeed, therapeutic doses of ABE did not significantly altered QT interval at the maximal steady-state concentration either in healthy volunteers and cancer patients.19,33
Pharmacokinetic interactions
Pharmacokinetic interactions occur when a drug affects another drug’s absorption, distribution, metabolism, or excretion. Less frequently, this DDIs are caused by physicochemical contrasts between two molecules, which alters the chemical structure of one or both drugs. 31
Absorption
Oral administration represents the most advantageous route for drugs consumption. However, several factors may interfere with the absorption of orally taken drugs, including concomitant medications, foods, or gastric pH.34,35
For instance, grapefruit juice and St. John wort may reduce the absorption of CDK4/6i and inhibit intestinal CYP3A4 activity, consequently reducing pre-systemic clearance of CYP3A4 substrates. Absorption-related interaction may also occur with calcium supplements, commonly prescribed to BC patients, which may block gastrointestinal uptake of some drugs, and with agents interfering with gastrointestinal motility and/or pH or affecting the microbiome. In some cases, complexes formation may inhibit absorption. 31
Proton pump inhibitors (PPIs) are among the most frequently prescribed drugs. According to previous evidence, PPI may interfere with PAL absorption, due to the pH-dependent solubility of this drug. 35 This interaction was supposed to determine decreased PAL efficacy and increased toxicity.35–37 Due to these observations, PAL capsule have been recently reformulated into tablets, whose absorption is not affected by gastric pH or PPI use. The existence of interference between PPI and RIB remains controversial, whereas it has not been reported for ABE. 38 However, it is possible that PPI interactions with CDK4/6i represent a class effect, thus affecting also RIB and ABE, but such evidence has to be proven.
Distribution
Distribution-related interactions may occur when two or more highly protein-bound drugs compete for the same binding sites on plasma proteins. Drug distribution is determined by bloodstream flow and by the binding to plasma proteins, such as albumin, albumin/globulin ratio (AG), lipoproteins, and immunoglobulins. 29 If two highly-bound to plasma protein agents are simultaneously administered, one can displace the other from its protein-binding site and increase the concentration of the unbound form. Unbound drugs are biologically active, while plasma protein binding limits the activity of the drug. Hence, displacement of bound drugs from blood components or tissue-binding sites is thought to increase the apparent volume of distribution of an agent. However, the effect of drug displacement is difficult to measure. 26
Generally, kinase inhibitors are moderately/highly bound to plasma protein and, as such, may be susceptible DDIs when co-administered with other protein-bound agents. Among CDK4/6i, PAL and RIB display a moderate protein bound, whereas the fraction of ABE and its active metabolites bound to plasma proteins is higher. However, none of the CDK4/6i seem to present clinically significant distribution-related DDIs. 6
Metabolism
Concurrent medications may alter drugs metabolism, potentially leading to clinically relevant effects.39,40 The most common metabolic DDIs include reversible inhibition, time-dependent inhibition and induction of drug-metabolizing enzymes, and stem from interactions with cytochrome P450 and/or P-gp.41,42
Cytochrome P3A4 inhibitors may decrease enzymatic activity, causing accumulation of the drug in the blood and enhancing toxicity. On the other hand, CYP3A4 inducers may speed up the production of substrates, decreasing drug plasma concentration, and eventually hampering efficacy. Metabolism-related DDI can be predictable to a certain extent, thanks to scientific data on drug metabolic and bio-transformation profiles. However, such information cannot always be applicable in the clinical practice, since they derive from in vitro or animal models, and do not take into account individual pharmacogenomic variations, enzyme kinetics, the contribution of multiple different drug-metabolizing enzymes, and drug clearance in humans. 43
Metabolic interactions represent the most frequent DDIs in patients treated with CDK4/6i. 7 While a description of PAL and RIB metabolism-related DDIs goes beyond the scope of this review, ABE metabolic interactions are detailed in the next paragraph.
Excretion
DDI affecting the excretion phase of a drug mainly involve its renal elimination. 44 In the kidney, interactions may occur not only because of competition during tubular secretion, but also for urinary pH alteration. 45 Nonsteroidal anti-inflammatory drugs may reduce renal excretion of concomitant administered agents. Such DDIs may cause up to 26% of all adverse drug events and are associated with a significant burden on the healthcare system through increased hospitalizations. 14 CDK4/6i are mainly eliminated in the biliary tract and therefore clinically relevant pharmacological interactions involving their excretion phase have not been reported thus far. 7
Pharmacokinetic model to predict DDIs
Clinical pharmacology proposes therapeutic drug monitoring (TDM) as a tool for individualized treatments that maximize a rational drug use. 46 With this approach, it is possible to calculate individualized dosing regimens by measuring drug exposure, pharmacological markers, or pharmacodynamic indicators in the patients’ body using quantitative pharmacological models based on the drug treatment window. For instance, Posada et al. 25 developed a physiologically based pharmacokinetic (PBPK) model incorporating ABE and its metabolites to predict the effect of strong and moderate CYP3A4 inhibitors and inducers.
The use of PBPK modeling to support dosing recommendations in regulatory submissions and prescribing labels has increased in recent years.47,48 Regulatory acceptance of such PBPK-driven recommendations increased concurrently, as the models become more robust and can simulate ever more complex scenarios. 25
ABE pharmacological interactions
Drug–drug interactions
In the clinical practice, many cancer patients take multiple medications for concomitant diseases. Therefore, exploring potential DDI using a drug interaction checker is strongly advisable before prescribing ABE. The interaction checker available online at Drugs.com (https://www.drugs.com/interactions.html) categorized DDIs as follows:
– Major: Highly clinically significant (avoid combinations, the risk of the interaction outweighs the benefit)
– Moderate: Moderately clinically significant (usually avoid combinations; use it only under special circumstances)
– Minor: Minimally clinically significant (minimize risk; assess risk and consider an alternative drug, take steps to circumvent the interaction risk and institute a monitoring plan)
Among the 286 drugs known to interact with ABE, 81 (28%) interactions are categorized as major, 205 (72%) as moderate, and 0 as minor. As shown in Figure 3, ABE displays the lowest number of drug interactions compared to PAL and RIB. In addition, both ABE and PAL have fewer major interactions compared to RIB.

Number and type of drug interactions according to the CDK4/6i.
ABE as a victim of DDIs
Inhibitors of cytochrome P450
Co-administration of ABE with CYP3A4 inhibitors is a relative contraindication, due to increased ABE plasma concentrations and ADRs risk. This means that oncologists should avoid the concomitant use of potent CYP3A4 inhibitors and ABE, or if needed, reduce the dose of ABE and carefully monitoring the patient for toxicities. 15
Antifungal agents (itraconazole, ketoconazole, posaconazole, or voriconazole) may profoundly interfere with the ABE metabolism. Indeed, systemic exposure to ABE may increase by up to 16-fold when co-administered with potent CYP3A4 inhibitors, such as ketoconazole. 49 In addition, itraconazole may increase the relative potency-adjusted unbound area under the curve (AUC) of ABE and its active metabolites by 2.2-fold.
Concurrent administration of the antibiotic clarithromycin or of the antiviral agent lopinavir/ritonavir 50 mg, single-dose ABE determines a 2.5-fold increase in the relative potency-adjusted unbound AUC of ABE and its active metabolites. 25
The moderate CYP450 3A4 inhibitors diltiazem and verapamil may increase the relative potency-adjusted unbound AUC of ABE and its metabolites by 2.4- and 1.6-fold, respectively. However, no dose adjustment is generally necessary for patients taking moderate or weak CYP3A4 inhibitors. 25
Cytochrome inducer drugs
Concomitant use of strong CYP3A4 inducers should be avoided due to the risk of decreased efficacy of ABE. Potent cytochrome inducers include antiepileptic agents, such as carbamazepine and phenytoin, antibiotics (rifampicin), and St. John’s wort. Co-administration of rifampicin and ABE decreased the plasma concentration of the latter by 95%, and unbound potency-adjusted plasma concentration of ABE plus its active metabolites by 77%. 50
ABE as a perpetrator in DDIs
Drug transporters
ABE exerts an inhibitory effect on several membrane transporters and can interfere with agents that are their substrates. 5 In fact, ABE and its active metabolites can inhibit organic cation transporter 2 (OCT2), multidrug and toxin extrusion protein 1 (MATE1), and MATE2K in the kidney. Interactions of ABE with clinically relevant substrates of these transporters, such as dofetilide or creatinine, may occur in vivo. For example, co-administration of metformin (substrate of OCT2, MATE1, and 2) and ABE (400 mg) determined an increase in metformin plasma exposure, not clinically significant. This effect was due to reduced renal secretion with unaffected glomerular filtration. 51 Therefore, no dose adjustments of ABE are necessary for patients with mild or moderate impairment of renal functions. Few data are available about ABE use in patients with severe renal impairment, end-stage renal disease, dialysis. Despite reports about safe use of ABE in dialyzed patients have been published, this drug should prescribed cautiously in case of severe renal function impairment. 52
In healthy subjects, co-administration of ABE and the P-gp substrate loperamide increased loperamide plasma exposure by 9%. However, this was not considered clinically relevant. Based on the inhibition of P-gp and BCRP observed with ABE in vitro, in vivo interactions with narrow therapeutic index substrates of these transporters, such as digoxin or dabigatran etexilate, may occur. 25
Drug–food interactions
ABE interactions with food and dietary habits are also worth to be considered. According to prescribing information, ABE can be administered with or without food, since food has only moderate effects on ABE pharmacokinetic. However, a high-fat diet may increase ABE bioavailability. 49 As reported in the FDA access data, ‘a high-fat, high-calories meal (800–1000 calories; 150 calories from protein, 250 calories from carbohydrate, and 500–600 calories from fat) administered to healthy subjects increased the maximum plasma concentration and AUC of ABE and its active metabolites by 26% and 9%, respectively.’ 19
Grapefruit juice may increase ABE plasma concentrations probably due to inhibition of CYP3A4-mediated first-pass, gastrointestinal metabolism, or hepatic CYP450.15,53 As a consequence, increased ABE exposure may enhance ADRs. In addition, grapefruit juice exerts their metabolic effect in a concentration-, dose-, and preparation-dependent fashion, with broad variability among brands. Indeed, high-dose or double-strength preparations may produce a potent CYP3A4 inhibition, whereas low-dose, single-strength formulations have typically demonstrated moderate inhibition. The clinical impact of these interactions is not definitively clarified.15,53
Nutritional counseling may be advisable for patients receiving ABE to reduce the risk of gastrointestinal side effects. In case of diarrhea, the banana, rice, apple, and tea (BRAT) diet should be suggested. 54
Interactions with complementary and alternative medicine
Complementary and alternative medicine (CAM) is widely used in cancer patients to aid recovery or treat symptoms associated with antineoplastic treatments. 55 Besides its potential effect on symptoms and outcomes, CAM may expose patient to the risk of interactions with antitumoral agents which are largely unknown.
The World Health Organization has recently recognized traditional Chinese medicine, which is of widespread use, especially in the far east. Therefore, many patients may take herbal treatments along with traditional medications, increasing the risk of DDIs. 56 For example, the active component of Astragalus membranaceus (i.e. astragaloside IV) seem to hamper tumor progression and metastatic potential and apparently enhances chemosensitivity in a BC rat model. 57 However, it interferes with ABE pharmacokinetics, increasing its systemic exposure through CYP3A4 inhibition. 56
Drug–gene interactions
Roncato et al. 58 reported a small-sized experience of patients who underwent clinical pharmacological counseling, including TDM, pharmacogenetics, and DDI analysis to support clinicians in managing CDK4/6i treatment for ABC. Three cases presented a CDK4/6i plasma level above the population mean value and were referred for toxicity. One patient presented a low functioning ABCB1 haplotype (ABCB1-rs1128503, rs1045642, and rs2032582), possibly causative of increased drug oral absorption and plasmatic concentration. Two patients showed underexposure to CDK4/6i, and one was referred for early progression. In one patient, a CYP3A5*1/*3 genotype was found to be potentially responsible for more efficient drug metabolism and lower drug plasma concentration. 58 More recently, the same group reported the results of pharmacogenomic assessment on large cohort including 230 BC patients treated with CDK4/6i. A significant correlation was present between the presence of polymorphisms in CYP3A4, CYP3A5, ABCB1, and ABCG2 and drug-related toxicities or the need for CDK4/6i dose reduction. 59
Other reports supported the relationship between ABCB1 homozygous polymorphisms (2677G>T/A) and increased ABE toxicity, due to higher concentrations of its active metabolites M2 and M20.60,61
Pharmacogenomic evaluation in clinical practice may support prescribers therapeutic choices. However, although pharmacological counseling could ultimately help increase the safety and efficacy profiles of CDK4/6i treatment, it seems still poorly applicable in the daily clinical practice.
Conclusion
The number of HR+/HER2− patients treated with ABE worldwide is destined to increase due to the widespread use of this drug. Hence, physicians should retain general knowledge about ABE pharmacological proprieties to fully exploit them in the clinical practice.
Compared to the other CDK4/6i, ABE displays some unique pharmacological features. First, ABE is administered on a continuous daily schedule, which allows a sustained target inhibition and a durable cell-cycle blockage.16,17 Differently from PAL and RIB, ABE has a strong binding rate to plasma proteins during its distribution phase, and this feature must be considered in case of patients with hypoalbuminemia. Additionally, ABE is the only CDK4/6i penetrating the BBB, and intracranial objective responses have been observed with this drug. 26 Lastly, ABE effect on tubular transporter in the kidney may lead to an increase in serum creatinine, which does not correspond to a renal function impairment. 51
When considering ABE pharmacokinetics, several pathophysiological should be also taken into consideration. For instance, body mass index and body composition may affect the distribution ABE – a highly lipophilic drug – and influencing its toxicity and efficacy.62,63 A pooled analysis of MONARCH2 and MONARCH3 trials showed a higher response rate and a lower incidence of neutropenia in lean patients compared to those overweight or obese. 62 Pre-existing impairment of liver and kidney function may impact as well ABE pharmacokinetic and should be incorporated into a comprehensive clinical evaluation. 6
Medical oncologist should also be aware of the potential DDIs and drug–food interactions related to ABE use. Indeed, a growing number of BC patients are exposed to polypharmacy, a condition that increase DDI risk, eventually leading to unexpected ADRs or diminished anticancer treatment efficacy.64,65 In line with this assumption, a recent report showed that concomitant use of multiple drugs was significantly associated with enhanced toxicities and reduced progression-free survival in a cohort of 173 Italian BC patients treated with ABE. 66
Several user-friendly online tools allow to freely check for DDIs in the clinical practice. Even though these instruments are generally reliable, there is no evidence of superiority for one of them over the others and several discrepancies have been found among these tools.5,6,67 Clinicians should be aware of these limitations and avoid to replace online interaction checkers with the technical data sheet of the drug. Additionally, online tools can be of little utility in case of complex or multiple drug interactions. In such instances, asking for a pharmacologist interventions may be the best option.
Of note, ABE displays significantly less DDIs compared to PAL and RIB. This unique feature makes ABE a manageable drug in the clinical practice. Also, fewer interactions likely translate into higher treatment adherence, which is of paramount importance to achieve and maintain disease response.
In conclusion, ABE represents an unprecedent resource for the treatment of HR+/HER− BC. Medical oncologists should be informed about the pharmacological characteristics and interactions of this drug in order to avoid preventable toxicities and maximize its efficacy.