
Abstract
The assessment of the safety of medicines for rare diseases during the development phase is often limited by the few data available from small numbers of patients. This also applies to a lesser extent during the postmarketing phase of the lifecycle of a medicine. By using all available sources of data for rare diseases drugs, and by carefully assessing these data, the most informed safety profile can be obtained. This should also allow a clear view of data that are not available at any given time point and facilitates planning of strategies to obtain data through appropriate postmarketing risk management. Although it is not always easy, there are possibilities to increase the speed by which data in the postmarketing period can be generated by better use of data from ongoing formal clinical trials, by early planning of drug or disease registries and leveraging the power of both disease patient support groups, which are often well established, and networks to facilitate international research, specifically in rare diseases. The future may offer approaches using personal medical monitoring data tools and ‘big data’ to further facilitate the availability of information and to determine the effectiveness and safety profiles of drugs used for rare diseases and thus allow the benefit/risk of these drugs to be optimized. These issues will be discussed here.
Keywords
Introduction
Major scientific advances, particularly in the area of biotechnology including monoclonal antibodies, antisense oligonucleotides, fusion proteins and others, have enabled pharmaceutical companies to produce drugs as replacement therapy in genetic disorders of metabolism or utilize interactions with novel biological targets for the production of new medicines. At the same time, incentives in terms of extended market exclusivity,1,2 reduced fees by medicines regulatory agencies, 3 cost recovery programmes and high prices which can be obtained for the so-called orphan drugs developed for the treatment of rare diseases have encouraged pharmaceutical companies to allocate more resources to this area. Further, development of new medicines for rare diseases has been enhanced by the establishment of a number of organizations conceived to facilitate information flow and research and development of rare disease therapeutics. These include the National Organization of Rare Diseases (NORD), the International Rare Disease Support Network (IRDSN) and the European Rare Disease Therapeutic Initiative (ERDITI), a coalition of patient organizations fostering interactions between academic institutions working on rare diseases and the pharmaceutical industry to bring new therapeutics to the marketplace. 4 These influences have resulted in increasing numbers of new treatments becoming available for previously poorly treated or untreatable rare diseases.
Interestingly, at the present time, there is no accepted universal definition of rare disease. From a medicines regulatory point of view, in the USA and the European Union (EU) orphan or rare diseases are defined as those that affect less than 200,000 individuals (prevalence <650/million population) and less than 5/10,000 (500/million) respectively. A systematic search for definitions related to rare disease from organizations in 32 international jurisdictions resulted in 296 definitions from 1109 organizations published by Richter and colleagues. 5 The average prevalence threshold across organizations within individual jurisdictions ranged from 5 to 76 cases/100,000 people. The global average prevalence from the definitions used in the different jurisdictions would be 40 cases/100,000 people. Within rare diseases, a subcategory of ultrarare diseases is often discussed. It has been proposed that a disease is considered to be ultrarare if it affects 1 patient per 50,000 people 6 (or fewer than 20 patients in a population of 1 million). In recent years, treatments for these ultrarare diseases have been investigated and a number of new therapies are now marketed. Initially the majority of treatments were enzyme replacement therapies for patients with genetic enzyme deficiencies such as those found in mucopolysaccharidosis or glycogen storage diseases. However, newer approaches, including chaperone therapies for enzyme deficiencies and treatments using antisense oligonucleotides for degenerative neurological diseases such as spinal muscular atrophy degenerative, are now available. It is likely that gene therapies will become available in the near future.
The efficacy and safety profiles of orphan drugs developed to treat ultrarare diseases are very difficult to determine. This is due to very few data or the lack of data available on the disease’s natural history and the use of the drug in humans. In particular, patient exposure is limited during the initial development phases and in most instances also at the time of approval. The small numbers of patients present particular problems in the investigation of the safety profiles of medicines used to treat these ultrarare diseases and this is further challenged by the fact that many of them are present in the paediatric population. It is therefore extremely important that the elements and procedures of safety monitoring plans for orphan drugs for rare diseases are carefully evaluated and based upon information available about the drugs from all sources, including chemistry, manufacturing and controls, nonclinical toxicology information, and any data from previous human experience for other indications other than that in the rare disease. If available, safety profiles of other approved drugs of the same class for other indications should also be used. 7
The various issues encountered in pharmacovigilance of medicines for the treatment of rare diseases and some of the possible solutions will be discussed.
Clinical development safety
During clinical development of drugs for ultrarare diseases, safety data, including adverse events, laboratory data, electrocardiograms and so on, are collected as is usual for any drug development programme. The limited size of a study population for an ultrarare indication, sometimes only tens or hundreds of patients, results in a small safety dataset at the time of drug approval. This means that only relatively commonly occurring adverse drug reactions can be detected. For example, according to the ‘Rule of 3’s’, the absence of an important adverse reaction affecting the safety of patients in a study population of 300 would only exclude, with 95% confidence, its true occurrence in 1/100 subjects. 8 In addition, developmental clinical studies in such ultrarare indications are often uncontrolled or comparative data are collected from a historical control group. Historical data are often used primarily to determine statistical differences in predetermined endpoints to assist with proof of efficacy of new medicines. This is particularly the case when published historical data are used. The usefulness of historical data to assist with the establishment of the safety profile of a new medicine can be enhanced if historical data are collected directly from patient records. The aim should be to collect as much clinical information as possible, including documented adverse events and laboratory data to be aggregated to match as closely as possible the safety data which are normally collected for any control group of a prospective clinical trial. However, the safety profile at the time of marketing, apart from common or even very common and predictable adverse reactions, is relatively poorly defined. This means that postmarketing pharmacovigilance to define the safety profile takes on particular importance.
Postmarketing pharmacovigilance
Spontaneous adverse drug reaction reporting
Spontaneous reporting of suspected adverse drug reactions occurring during the marketed use of medicines has been the mainstay for the detection of unknown adverse drug reactions since it was introduced over 50 years ago. It is most useful for the detection of very rare syndromes not occurring in usual disease states (e.g. occulomucocutaneous syndrome with practalol, Achilles tendon rupture with quinolone antibiotics) and, as originally conceived, requires both adequate observation and implicit causal association as judged by a reporting physician. In more recent years, reporting of medical events occurring during treatment with a medicine, not only by a wider group of healthcare professionals, but also by patients, has meant that, whilst more adverse event data are collected, causal association with drug treatment cannot always be implied with the same level of certainty. This has been further complicated by the solicitation of adverse events through direct contact with patients by pharmaceutical companies using so-called patient support programmes and the requirement in the EU to report adverse events collected during market research activities. However, a well documented individual case report of an important suspected adverse reaction can be useful to identify unknown adverse drug reactions in rare diseases, but it requires strenuous efforts to gather comprehensive information about the patient and their treatment leading up to the event to establish causality, including temporal relationship, dechallenge and rechallenge, potential risk factors, alternative explanations and consideration of plausibility. Additionally, for some drugs used to treat rare previously lethal diseases of infancy and childhood, for example inborn errors of metabolism and storage diseases, the patient populations being treated are very sick and have a high incidence of adverse events related to the underlying disease as alternative explanations, making causal association difficult to determine. In addition, a treatment that prevents a previously observed fatal outcome in a childhood disease creates a new population of survivors. These survivors will have a course of disease which will never have previously been seen. As a patient grows older and the disease progresses, a reported new (perhaps serious) adverse event could be the consequence of the disease itself rather than an adverse drug reaction. There will be no background incidence to determine the likelihood of the occurrence of such an event in the absence of drug therapy.
As the numbers of reports of adverse events collected from the market have increased and qualitative examination of all individual reports has become untenable, an alternative method of data mining to detect possible new adverse drug reactions has been developed. 9 This method uses statistical analyses of drug adverse event databases to detect signals of possible adverse reactions (quantitative signal detection). Signals of possible adverse reactions are then investigated to determine if a signal can be confirmed or refuted as a new unknown adverse reaction. 10 The small patient exposure for medicines used to treat rare diseases and the resulting low numbers of reports of spontaneous suspect adverse drug reactions, as well as the likelihood of rare but often common concomitant pathologies, particularly in many rare genetic diseases (confounding by indication), makes quantitative signal detection both inefficient and insensitive for the identification of signals of possible adverse reactions. Therefore qualitative methods and the adoption of alternative proactive approaches (e.g. noninterventional safety studies to collect data during the postmarketing period) are more useful to define the safety profile and to assist with the surveillance of the benefit/risk profile of drugs used to treat rare diseases.
Postapproval safety studies
For many years in Japan, pharmaceutical companies have been legally required to undertake postmarketing epidemiological studies to collect safety information after first approval of a medicine and there are guidelines for operational implementation of such studies. 11 Legislation in the EU and the USA, as well as increasing numbers of other countries, now also allows regulators to impose requirements for pharmaceutical companies to undertake further studies or additional risk minimization measures, usually at the time of the first marketing approval. These become part of a formal EU risk management plan or a US Food and Drugs Administration risk evaluation and mitigation strategy. The exact nature of the studies requires agreement between the pharmaceutical company and the regulator, and must consider not only the most appropriate design of the study but also the practicalities of its implementation.
Formal clinical trials
It is usual when medicines are newly approved that there are still ongoing open-label studies which are often extensions of earlier studies, from which data have been used for marketing approval. Many regulators encourage companies to continue these studies in order that the long-term safety profile of a well defined cohort of patients can be studied. Such data can be useful to determine whether relatively common new adverse reactions due to delayed or cumulative effects of a new medicine occur, and an absence of such can provide early reassurance of the long-term safety. In addition, other clinical trials, often in a paediatric population for the same indication or for totally new indications in adults, are undertaken by companies. Safety data from these trials can be used to bolster patient exposure to better define safety profiles, although the limitations of data from formal trials for determination of a safety profile during marketed use remain. Safety data from clinical trials therefore are of importance not only in the preapproval stages but also in the postapproval stages of the lifecycle of a medicine.
Epidemiological studies
Registries and observational studies
A typical postmarketing epidemiological study for the first medicine to treat a rare disease is either a disease or a drug registry. A registry is an organized system that uses observational methods to collect uniform data in a population defined by a particular disease condition or exposure. A registry can also be used as a data source within which further studies can be performed or can include substudies such as those recruiting pregnant, elderly or paediatric patients. After patients are recruited into the registry they can be followed over time and therefore incidences of adverse events can be accurately calculated. Many of the first therapies for rare diseases have used a drug registry to obtain safety data during use of a drug on the market. If properly designed, a registry can also be used to offer insight into the exact population treated on the market (drug utilization). When designing a drug registry, it is important to define its objective and to also consider whether data can be enriched with data on outcomes, confounding variables and effect modifiers obtained from a linkage to an existing database, such as national cancer registries, prescription databases or mortality records. A drug registry is most useful when a specific safety outcome needs to be studied. A good example of this is the European Tracleer (Bosentan) registry 12 (albeit in a rare rather than an ultrarare disease). This medicine is used to treat patients with primary and some secondary forms of pulmonary hypertension and clinical trials demonstrated a risk of liver toxicity. The registry was used to obtain data on the exact incidence of liver toxicity during marketed use of the drug and also allowed investigation of any subgroups of patients at risk of this particular event.
When the requirement of pharmacovigilance is to better define the overall safety profile during marketed use and to identify important unknown adverse reactions, as is the case with most medicines for ultrarare diseases, one problem of a drug registry is the lack of data from a control group not treated with the particular drug under investigation. Without these causal associations between reported adverse events and administration of a treatment can be difficult to determine. For this reason, regulators often request that data be collected on patients not receiving treatment with the particular drug under investigation. This would then require a comparison of cohorts receiving or not receiving treatment with a particular medicine for a disease and to some extent such a registry becomes a disease registry rather than a drug registry. Pharmaceutical companies marketing enzyme replacement therapies for lysosomal storage diseases used disease registries to obtain safety data and effectiveness data during marketed use of their newly approved therapies. 13
Guidance on the design of such epidemiological studies has been provided in the EU Good Pharmacovigilance Practices Module VIII. 14 Standards for the collection and analysis of data usually follow the good pharmacoepidemiological practice guidelines of the International Society for Pharmaceutical Engineering. In addition, codes of conduct for transparency and independence of such studies have been produced by the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance, 15 among others.
There are many challenges faced in the design and implementation of observational registries. For rare and ultrarare diseases patient numbers remain limited, even during marketed use, and therefore recruitment of adequate patient numbers is difficult. In addition, sometimes healthcare professionals are unwilling to take part in these studies due to the limited compensation for their participation, and also because of concerns about prescribing often expensive drugs used for the treatment of rare and ultrarare diseases. Further, although a global study would be the most efficient approach for recruitment of adequate numbers of patients, because of differences in healthcare practice and healthcare provision as well as regulations governing epidemiological studies, it is often not possible to provide for such variations within a single common protocol. Given this, global studies/registries are relatively rare. More usually there are a number of local registries which recruit patients with slightly differing protocols but with some core data collection tools which allow pooling of data (such as baseline demographic data and safety data) for analysis. Such an approach also allows for different protocols to contain some substudy investigations that can allow local publication, which is an added incentive in the recruitment of physicians to undertake the study. For many rare diseases, however, the existence of already established country- or continent-specific research groups can provide a pharmaceutical company with a ready-made group of potential recruiters and the formal sponsoring of an epidemiological study by such groups can assist with investigator recruitment and also adds academic credibility to a study. In addition, it is possible to discuss with regulators the approval of a Dear Health Care Professional letter which can ask physicians to support and become investigators in postmarketing studies of a newly approved medicine. Rare diseases often have associated patient support groups and these are another resource which can be used to encourage patients to volunteer for participation in the registry studies and can assist with retention of patients within the registry once recruited. It is becoming more common that protocols first use direct contact with patients to collect information (patient-reported outcomes) and important information collected in this way is then further verified by patients’ healthcare professionals.
It is undoubtedly true that the establishment of a disease or drug registry is most difficult in diseases for which little or no prospective epidemiological research has been undertaken. This often applies to the drug registries for the first treatments approved for rare diseases. In addition, for first treatments in rare diseases, it is considered unethical once a treatment is available not to prescribe it to all patients and therefore an untreated control group is no longer available within the registry. For this reason, in order to enrol patients in a control group, it is important, if possible, to also implement registries to include countries where approval or general availability of new medicines is usually delayed through prolonged marketing approval processes or reimbursement negotiations.
When research groups for epidemiological research of treatments of a rare disease have been established, it is possible to leverage their experience for data collection with newer treatments. Such approaches also allow comparisons of effectiveness and safety profiles between different treatments. For this reason, drug regulators often request that pharmaceutical companies marketing different treatments for the same disease combine their resources to sponsor single disease registries collecting data on different medicines. Whilst in theory this would be the most efficient approach, commercial sensitivities can sometime hinder such collaborations. A good example of such a collaboration is the Antiretroviral Pregnancy Registry. 16 However, this is in itself a relative rarity and its establishment was assisted by pharmaceutical companies and prescribers acknowledging the urgent need to determine the effects of different antiretroviral treatments on pregnancy outcome for a disease with a large population of still fertile patients. The successful collaborative registries dedicated to the investigation of biologics used to treat rheumatological diseases are a further example. One of the first and best known is probably the UK British Society of Rheumatology Registry, 17 which monitors the effectiveness and safety of antitumour necrosis factor drugs. However, similar ones now exist in many countries and have provided invaluable data to assess the effectiveness and safety of new medicines, in particular to treat rheumatoid arthritis.
Healthcare databases
Information available in databases of healthcare records are a ready-made source of both beneficial and adverse effects associated with diseases and treatments. Research conducted over recent decades by epidemiologists using such databases has demonstrated their utility in the investigation of adverse drug reactions. The data can be used to analyse the detection of signals of potential new adverse drug reactions and formal adequately statistically powered epidemiological studies can be used to confirm or refute potential safety issues hypothesized to be causally associated with medicines. Classical comparative cohort studies or more specific case control studies have been undertaken using databases which contain actual patient medical records, most commonly the UK General Practice Research Database (GPRD) (now UK Clinical Practice Research Datalink (CPRD)), but many others have been used, in particular Canadian Provinces Health Care databases. Other investigations have used administrative databases which contain records of patients’ medical history used for payments and other financial functions associated with the provision of patient care. These are mainly based in the USA, with the publicly funded Medicaid and Medicare, and the privately funded Kaiser Permanente databases perhaps the best known. Despite their proven value in the investigation of safety of medicines used for more common diseases, the size of each of these databases has meant that the numbers of patients with rare diseases have been too small to be useful for studies of medicines used for these diseases. In response to the FDA Amendments Act of 2007, in May 2008 the FDA launched the Sentinel Initiative which aimed to combine data from a number of different database sources which could be used as a resource for improved signal detection and signal investigation of adverse drug reactions. 18 This initiative is now reaching early maturity and records of over 190 million patients are reported to be available. These numbers offer the opportunity that adequate numbers of patients with rare diseases will be represented to allow meaningful investigations of safety of medicines prescribed for these diseases. There are other initiatives to combine data from different sources, one of which is the Global Research in Paediatric (GRiP) network of excellence, 19 sponsored by the European Commission. This project aims to facilitate the development and promote the availability of medicines specifically for children and includes initiatives to improve availability of data for pharmacoepidemiology studies of medicines prescribed to children. As many rare diseases are genetic in nature and often present in childhood, global collaborations such as GRiP may eventually provide additional data resources for investigation of safety issues of medicines used in children with rare diseases. Although not yet fully available, these combinations of patient data from multiple sources could be particularly promising in the future for the study of rare diseases and the medicines used to treat them.
Effectiveness data
Although pharmacovigilance has classically in the main concerned itself with safety data, the primary motivation of the activity is to minimize risk and thereby maximize benefit/risk of medicines. In recent years it has been recognized that collection of not only safety information but also effectiveness information from marketed use of medicines is required to more accurately assess benefit and risk. This has taken on greater significance since the introduction of accelerated marketing approvals of medicines in the USA and conditional approvals for new therapies for diseases with unmet therapeutic needs in the EU. Many conditions with such an unmet therapeutic need are rare diseases. Medicines approved for marketing under these earlier market access initiatives often have mandatory requirements for further studies which include assessment of efficacy and effectiveness, as well as safety. Classical clinical trial methodologies or epidemiological methodologies can be used. This means that epidemiology studies, previously only collecting safety data and with the remit of safety and epidemiology departments, now often also collect data on effectiveness, particularly for medicines to treat rare diseases. The design of such studies requires close collaboration among not only pharmacovigilance and epidemiology personnel but also clinical disease experts and experts in the assessment of patient outcomes during marketed use of medicines. This is a rapidly developing area and is likely to become the standard for pharmacoepidemiology studies of medicines, particularly for rare diseases for which the availability of patients for formal clinical trial studies is limited.
The future
Although most new therapies for rare diseases are currently classical small molecules or biologics, future therapies are likely to use more innovative approaches, such as gene therapy as well as tissue and cell-based therapies. These will pose new potential safety issues and will require even closer collaboration than often currently occurs between clinical pharmacovigilance and manufacturing in order to monitor the safety of medicines. In addition, it is becoming more common for prescription of medicines to require personal genotyping in order to assess effectiveness (for example, migalastat for the treatment of Fabry disease 20 ) and adverse drug reactions, particularly allergic reactions (e.g. abacavir). 21 Future pharmacovigilance investigation is therefore likely to require collection of such information and the linking of data from various sources, possibly including biobank data, will require new approaches as well as new methodologies for analysis and assessment of data. The use of social media as well as remote monitoring of patient health with personal monitors to collect safety and effectiveness information are also being widely discussed. 22 Whether these potential new sources of data will assist with the assessment of safety and benefit/risk of medicines or simply create more noise through the collection of poor quality information is yet to be proven. However, it is clear that improved and bigger data sources as well as improved methodologies for data collection, as long as they are of good quality and can be verified, from these sources could assist with the assessment of safety of medicines used in rare diseases.
Conclusion
As is usual with rare diseases, there is a paucity of data at the time of marketing approval; as such, it is important to use data from as many sources as possible to assess the safety profile of drugs used for these diseases (Figure 1). These data can be used in the development of a risk management plan to propose postmarketing studies, which will provide further data to contribute to the emerging safety profile and often the effectiveness profile during marketing use of such drugs. Similarly in the postmarketing phase, data from all available sources need to be used and assessed to update safety and effectiveness profiles as they emerge. This would include any data that become available through technical developments of methods for the collection of medical data, such as personal medical monitoring. If appropriate, updates to information provided to healthcare professionals and patients will need to be implemented (Figure 2) to ensure that at all times the applicable benefit/risk of a medicine is being communicated.
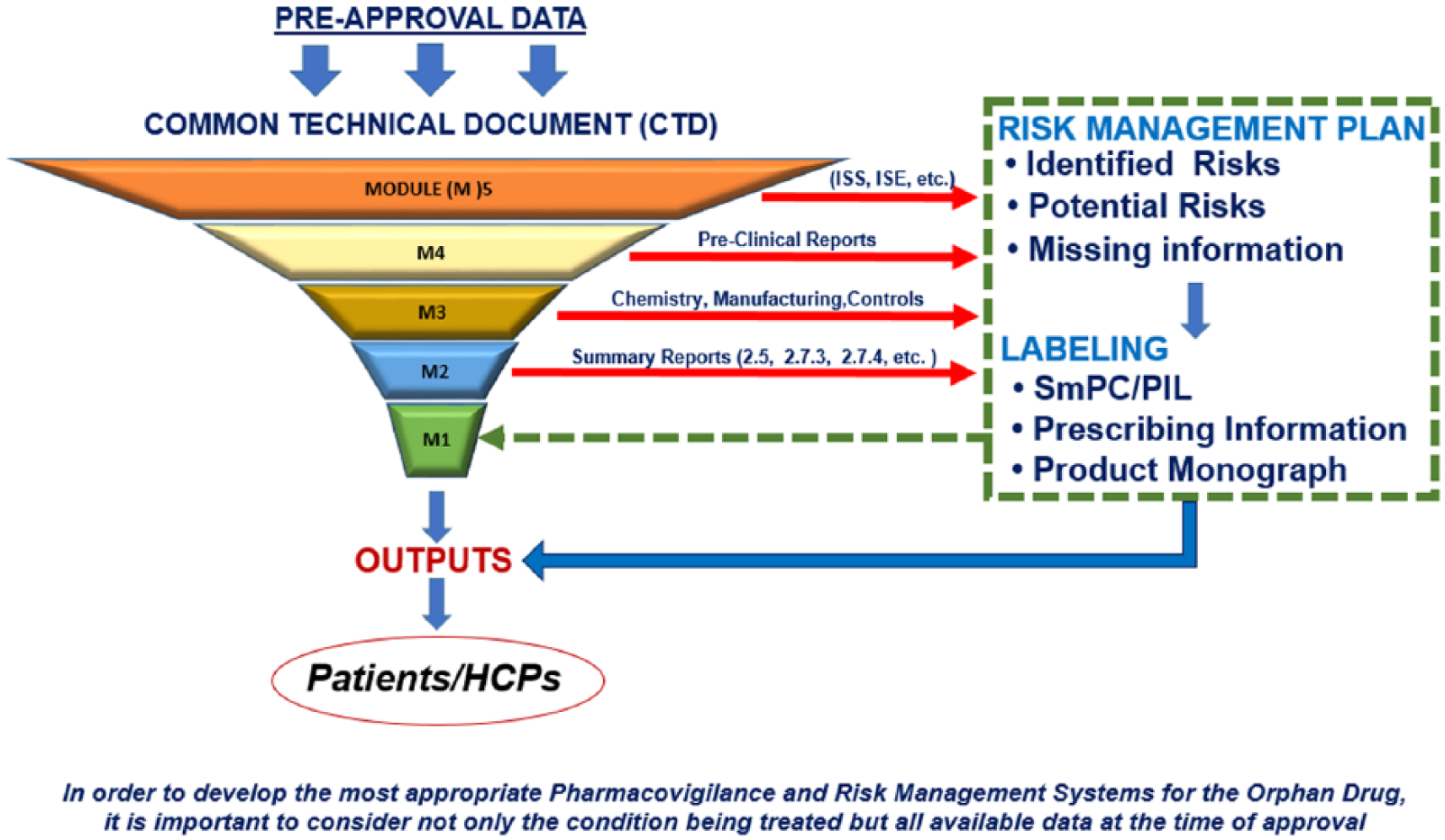
Development of pharmacovigilance and risk management systems for orphan drugs. HCP, healthcare professional; SmPC, Summary of Product Characteristics; PIL, Patient information leaflet; ISS, Integrated Summary of Safety; ISE, Integrated Summary of Efficacy.
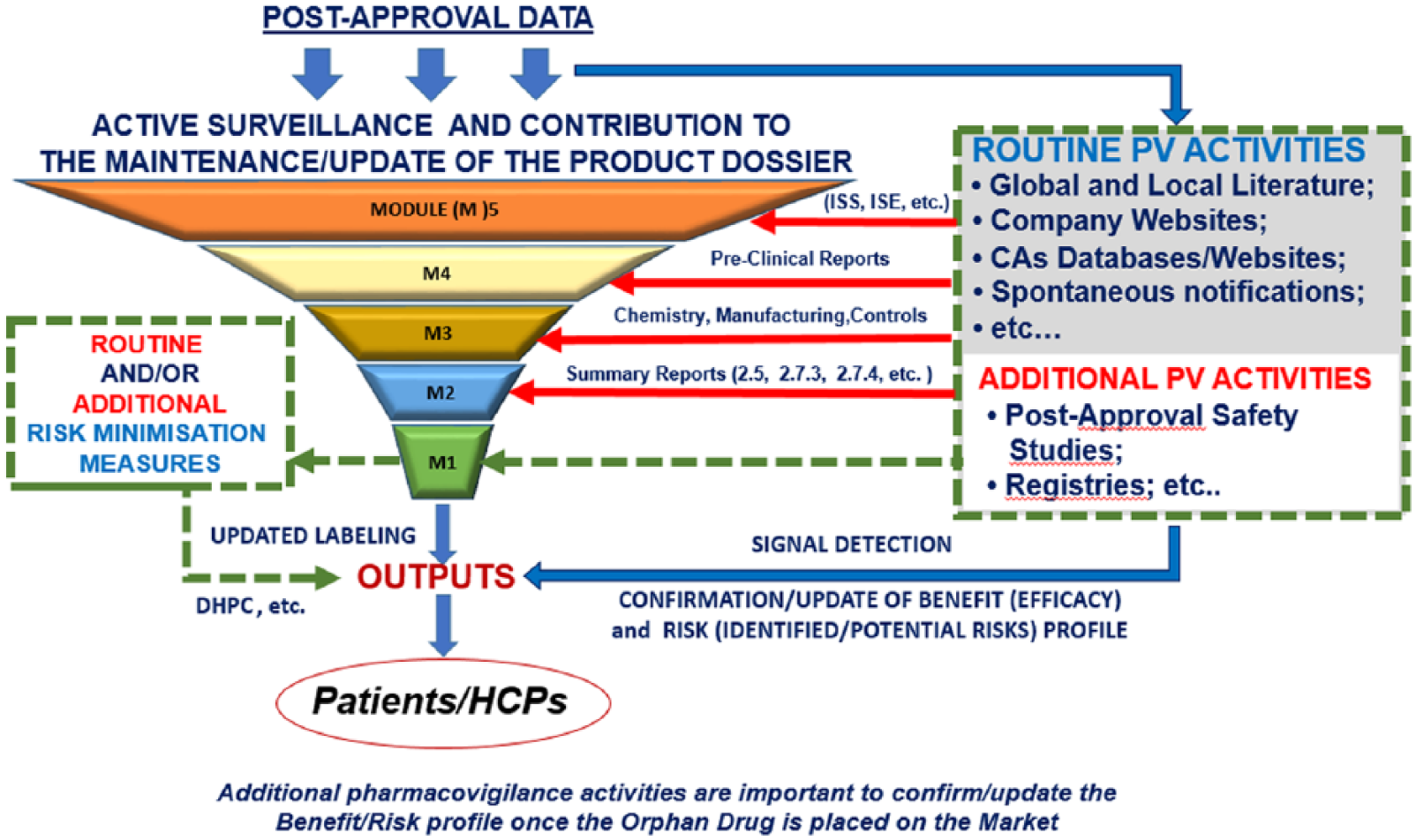
Surveillance of orphan drugs after approval. HCP, healthcare professional; PV, pharmacovigilance; DHPC, Dear Health Care Professional Communication; ISS, Integrated Summary of Safety ; ISE, Integrated Summary of Efficacy; CAs, Competent Authorities.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.