
Abstract
European Directives and Regulations introduced between late 2010 and 2012 have substantially overhauled pharmacovigilance processes across the European Union (EU). In this review, the implementation of the pharmacovigilance legislative framework by EU regulators is examined with the aim of mapping Directive 2010/84/EU and Regulation EC No. 1235/2010 against their aspired objectives of strengthening and rationalizing pharmacovigilance in the EU. A comprehensive review of the current state of affairs of the progress made by EU regulators is presented in this paper. Our review shows that intense efforts by regulators and industry to fulfil legislative obligations have resulted in major positive shifts in pharmacovigilance. Harmonized decision making, transparency in decision processes with patient involvement, information accessibility to the public, patient adverse drug reaction reporting, efforts in communication and enhanced cooperation between member states to maximize resource utilization and minimize duplication of efforts are observed.
Keywords
Introduction
Directive 2010/84/EC [The European Parliament and the Council of the European, 2010a] and Regulation [European Commission (EC)] No. 1235/2010 [The European Parliament and the Council of the European, 2010b] overhauled the European Union’s (EU) pharmacovigilance framework established by Directive 2001/83/EC [The European Parliament and the Council of the European, 2001] on the community code relating to medicinal products for human use (hereinafter referred to as the Directive) and Regulation (EC) No. 726/2004 [The European Parliament and the Council of the European, 2004] laying down community procedures for the authorization and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency (EMA) (hereinafter referred to as the Regulation) [Borg et al. 2011]. The amendments to the Directive and the Regulation became effective from July 2012 and impacted the authorization requirements for a marketing authorization (MA) [introduction of a pharmacovigilance system master file (PSMF) instead of a detailed description of the pharmacovigilance system, as well as a risk management plan (RMP) becoming a requirement for all new products; postauthorization measures were also enhanced with postauthorization safety studies (PASS) and postauthorization efficacy studies also becoming legally binding]. Furthermore, the EMA [for centrally authorized products (CAPs)] and national competent authorities (NCAs) [for nationally authorized products (NAPs)] need to evaluate the effectiveness of risk minimization measures (RMMs) through scrutiny of company reporting on its RMMs and measurement of drug utilization studies and health outcomes for key benefit–risk issues. The new EU legislation also introduced clarity in the oversight by the authorities of noninterventional studies: one member state is responsible for national oversight, while when more than one member state is involved then the EMA and its Pharmacovigilance and Risk Assessment Committee (PRAC) has the oversight of both protocol agreement and results assessment. In addition the new legislation provided for the development of an EU database listing all medicinal products authorized in the EU. This database will be populated by MA holders (MAHs) and lists for products subject to ‘additional monitoring’ which are labelled as such in the package leaflet will be compiled. With respect to adverse drug reaction (ADR) reporting, the ADR definition has been simplified to capture all noxious and unintended effects [including medication errors (MEs) resulting in harm] for reporting purposes. A shift in the definition has been made to allow the collection of information from all possible and available sources that will consequently lead to the development of an increased pool of information, from which relevant signals and useful information can be extracted and analysed. While patients and healthcare professionals continue to report exclusively at national level, patient reporting of ADRs is now a right of citizens in all EU member states. For the pharmaceutical companies, reporting suspected ADRs has been directed to EudraVigilance (the EU database of suspected ADRs) rather than to NCAs and thus creating a European pool of safety information. The EMA has been mandated to monitor literature (the EMA will monitor a selected list of substances and only in the scientific databases, while Marketing Authorisation Holders (MAHs) have to screen substances not on the list and screen local publications) for individual case reports of suspected ADR issues and to enter these into EudraVigilance and make them available to the pharmaceutical companies. The European Medicines Agency (EMA) is also mandated to report all European Union Adverse Drug Reactions (EU ADRs) regularly to the Uppsala Monitoring Centre of the World Health Organization. With respect to signal detection, this activity is now legally mandated and carried out for all products, where the EMA leads for Centrally Authorised Products (CAPs) and NCAs lead for NAPs with support from the EMA. Changes to periodic safety update reports (PSURs) according to International Conference on Harmonisation (ICH) E2C(R2) and good pharmacovigilance practice (GVP) Module VIII have also been introduced where these have become an evaluation of the benefits and risks of the product and are based on all cumulative data (rather than interval data as before). A periodic benefit–risk evaluation report is being generated with a view to examining the benefit–risk ratio throughout the lifecycle of the medicinal product. Within the EMA, a new committee PRAC has been established focusing on Periodic Safety Update Reports (PSURs), referrals, Post Authorisation Safety Study (PASS), risk management, signal detection and evaluation and effectiveness of Risk Minimisation Measures(RMMs). PRAC can be viewed as the major advisory board on aspects of safety for Committee for Human Medicinal Products (CHMP), a European Medicines Agency, when it relates to centrally authorized products or applications submitted through the centralized procedure and for National Competent Authorities(NCAs) through their coordination group; Pharmacovigilance and Risk Assessment Committee (PRAC) recommendations are sent to CHMP or the coordination group for adoption as appropriate.
At preauthorization stage, safety assessment focuses on the totality of the data collected up to the point of the application for MA and it can be considered as an exhaustive assessment. The amendments in the legislation shifted part of the assessment burden from the preauthorization stage and placed emphasis on the surveillance in the postauthorization setting. Several medicinal products have been withdrawn due to safety issues over the past 15 years and these are summarized in Table 1. These can be considered the main drivers for the change in the regulatory approach and the enhancement of the pharmacovigilance procedures and activities. Other reasons for the change in the legislation included the realization that all the information for the safety of a medicinal product cannot be known at the time of authorization; limitations and uncertainties in the data submitted and acceptable lack of knowledge at the time of authorization that cannot be completed unless the product enters the markets; diversity in the population of patients who will use the medicinal product that cannot be foreseen at the time of authorization. Finally the need to increase the pool of information including all available sources such as patients and literature reports is also an important driver of the change in the legislation.
Examples of drugs withdrawn for safety reasons in all European Union member states between 2000 and 2011 (modified from McNaughton et al. [2014] and Post-Licensing Directorate [2011]).
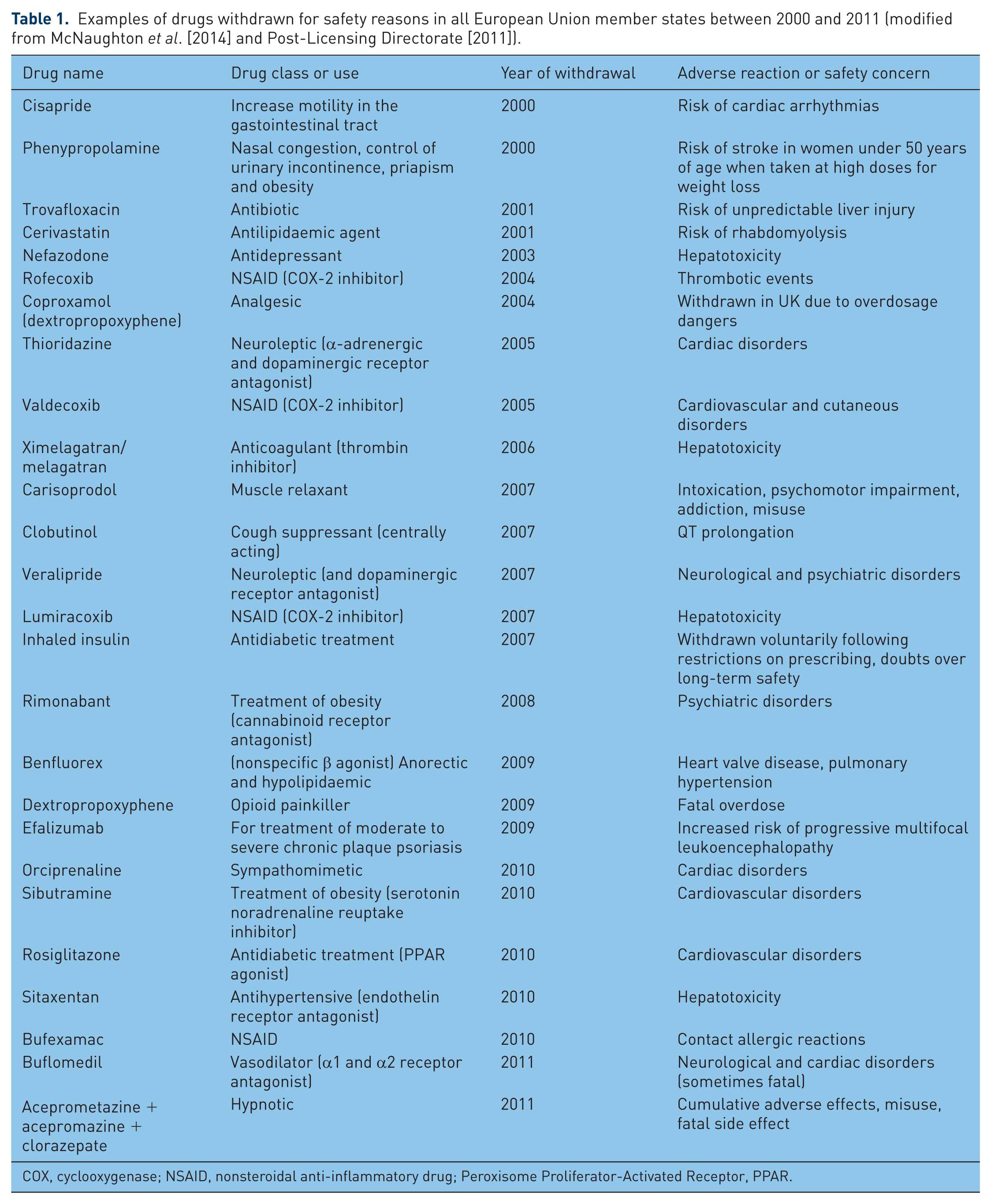
COX, cyclooxygenase; NSAID, nonsteroidal anti-inflammatory drug; Peroxisome Proliferator-Activated Receptor, PPAR.
It is easily understandable that the legislative change meant a significant increase in workload within pharmacovigilance departments across all sectors, with some postulating that the industry expected a 2030% increase alone in case volume [Borg et al. 2011; Garattini and Bertele, 2011; Tanti et al. 2013]. In this review, we examined the implementation of the pharmacovigilance legislative framework by EU regulators with the aim of mapping out whether the Directive and Regulation are strengthening and rationalizing pharmacovigilance in the EU.
Literature search methodology
To identify relevant literature, we performed an EU Council Public Register, EU Parliament Public Register, EU Committee of Regions Public Register, EUROPA (EC), Eur-Lex, and European Medicines Agency search (December 2010–September 2014). We used the search term ‘pharmacovigilance’ and included all documents written in English.
Results and discussion
Legislative updates
In 2012, the EC adopted legislative proposals (a Directive and a Regulation) to amend once again the European legislation on pharmacovigilance [The European Parliament and the Council of the European, 2012a, 2012b]. This followed the issue with the ‘mediator case’ in France. Mediator was a medicinal product prescribed to patients with diabetes and obesity. A number of deaths associated with the use of this product from cardiovascular events were observed by health professionals and not through the usual pharmacovigilance means. The case of ‘mediator’ has had consequences in the way pharmacovigilance is conducted in one member state and in Europe as a network through changes to the legislation. To avoid such scenarios being repeated, the EC ‘stressed’ (tested) the recently amended EU’s pharmacovigilance regulations (Directive 2010/84/EU) with respect to situations that could potentially mimic the ‘mediator’ scenario. There is also the well known story of thalidomide that was used for morning sickness in pregnant women and its administration led to severe malformation of the limbs in the infants born to these women. The case of thalidomide was the main reason for the strengthening of the legislation and establishing the regulation of medicines globally in the 1960s. It is considered as the first case that introduced strict regulations and initiated rigorous assessment of all safety information available. Another known case that received a lot of publicity and criticism internationally is the case of rofecoxib, a cyclooxygenase-2 inhibitor (coxib), which is a nonsteroidal anti-inflammatory drug removed from the market due to safety concerns (increased risk of serious heart disease, heart attack and stroke associated with long-term, high-dosage use).
On the basis of this and other previous safety withdrawals in pharmaceutical history (please see Table 1), gaps in the legislation across all regions of the world were identified. Specifically, EU legislation issues were identified and addressed through Directive 2012/26/EC whereby harmonized EU-wide triggered arbitration procedures have become automatic for the most important issues. As a consequence, member states do not act unilaterally when it comes to issues of drug safety [The European Parliament and the Council of the European, 2012a, 2012b]. The Directive has been applicable to all member states from 28 October 2013. In 2012, the EC published an Implementing Regulation (referred to as the CIR), setting the minimum requirements for the quality systems of both MAHs and NCAs for the performance of pharmacovigilance activities [European Commission, 2012]. This CIR is the standard to which pharmacovigilance audits on pharmacovigilance systems are implemented and thus considered by the authors as of critical importance for all actors in the sector. Under the new legislation, uncertainties in safety as in the case of coxibs can now be handled proactively by a detailed RMP capable of identifying potential risks, efficient RMMs and by allowing additional postauthorization safety studies, while triggering a referral procedure and as a consequence a thorough review of all available data is also considered as a suitable option. Interestingly, only one withdrawal of a marketed product has been observed up to April 2015 since the implementation of the new legislation in 2012.
The primary concern identified by the Fraunhofer report in 2006 [Biihrlen et al. 2006] that EU regulators were acting in disharmony when taking regulatory action on safety issues across all medicinal products irrespective of the approval procedure (CAPs or NAPs) is considered to be resolved.
In 2014, the EC adopted a Regulation 658/2014/EC to introduce fees for pharmacovigilance activities at the level of the EMA. Fees have been introduced for referrals, PSURs and imposed noninterventional safety study protocol evaluations. These fees will be used largely to compensate the rapporteur teams in the NCAs of the member states who will perform the assessments for the PRAC. In addition, an annual fee payable to the EMA for national authorized products for information technology systems and databases, and signal detection activities has also been introduced. Annual fees to cover these mentioned activities of the EMA apply from January 2015. The EMA will start charging from 1 July 2015 [The European Parliament and the Council of the European, 2014].
The PRAC
The PRAC was set up in July 2012 and meets every month at the EMA. Analysing the monthly agendas of the PRAC on the EMA website (www.ema.europa.eu), it is interesting to note that since September 2012, the items for discussion grew steadily from 17 product-related issues and not procedural aspects (September 2012), to around 103 (January 2013), peaking at 222 (September 2014) and settling at around 138 (January 2014 to December 2014). Interestingly, the following frequency sequence of PRAC agenda item topics have been recorded: RMP (36.7–42.3%), PSURs (26.6–34.6%), signal detection (7.4–14%), CHMP safety issues (5.3%), PASS (3.4–17.6%), Article 31 referrals (i.e. risk–benefit referrals; 3.3%), pharmacovigilance inspections (2.2%), Coordination Group for Mutual Recognition and Decentralised Procedures (human) (CMDh) safety issues (1.4%), Article 107 referrals (i.e. urgent safety procedures; 1.3%), Article 5(3) referrals (executive director or member state referrals on issues of interest; 0.16%). From July 2012 to March 2015, there have been 8 Article 107i referrals, 8 Article 20 referrals and 23 Article 31 referrals. The results indicate that the PRAC workload is considerable and that the member states (through assessors) and the EMA (through scientific administrators) have responded to the mandate directed by the EU pharmacovigilance legislation. Table 2 is indicative of the PRAC activities, with examples of substances and medicinal products, and the outcome.
Examples of Pharmacovigilance Risk Assessment Committee (PRAC) activities for certain active substances between September 2014 and February 2015 (6-month period).
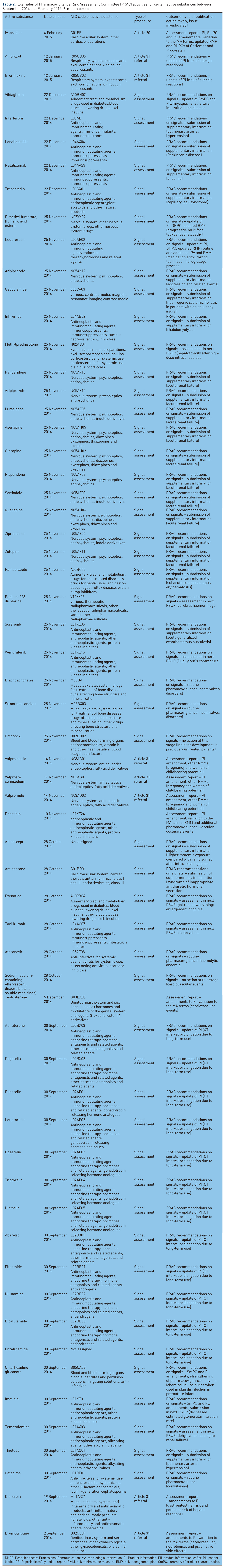
DHPC, Dear Healthcare Professional Communication; MA, marketing authorization; PI, Product Information; PIL product information leaflet; PL, patient leaflet; PSUR, periodic safety update report; RMM, risk minimization measure; RMP, risk management plan; SmPC, summary of product characteristics.
Good vigilance practices
A key deliverable of the Directive is development and implementation of a guideline on GVP. These practices constitute a set of measures with the scope of regulating the performance of pharmacovigilance in the EU. This guideline is applicable for MAHs and EU regulators (for all medicinal products: NAPs and CAPs) and is divided into chapters that fall into two categories: modules covering major pharmacovigilance processes; and product- or population-specific considerations (the latter are still under development). GVP Modules I–XVI cover major pharmacovigilance processes and are available on the EMA website (www.ema.europa.eu). Effectively, the GVP modules replace Volume 9A of The Rules Governing Medicinal Products in the European Union – Guidelines on Pharmacovigilance for Medicinal Products for Human Use [European Commission 2008]. For convenience, we have summarized the key elements that the GVP modules deal with in Table 3.
Good pharmacovigilance practice (GVP) modules.

EMA, European Medicines Agency; EU, European Union; MAH, marketing authorization holder; NCA, national competent authority; PSUR, periodic safety update report.
Periodic safety update reports
The recently published CIR (EU) No. 520/2012 [European Commission, 2012] has set the format and structure of PSURs, while more information on details and guidance for the submission of PSURs in the EU, data lock points and the frequency of submission are provided in GVP Module VII ‘Periodic safety update report’ (www.ema.europa.eu). The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use has developed an ICH harmonized tripartite guideline for the periodic benefit–risk evaluation report E2C(R2) that provides guidance on how periodic benefit–risk evaluation reporting on marketed products among the ICH regions should be formatted by MAHs for the evaluators and agencies. Importantly, the structure of PSURs has changed to an integrated benefit–risk analysis for authorized indications of the active substance covered in the PSUR. Thus it is expected that MAHs characterize both the risks and benefits and then critically appraise the uncertainties thereof. MAHs can now submit PSURs electronically to the EMA only as member states will have access to these submissions through a repository (available in 2015). Single assessments are carried out by drug substance in a procedure where all MAHs for products containing a particular substance submit PSURs at the same time for assessment at the PRAC (although PSURs for generic products are not routinely submitted). The PSURs assessed by the PRAC will cover any mix of CAPs and NAPs (including through the mutual recognition and decentralized procedures). The continuous circle of collecting data, reporting and evaluating them reassures us that the dynamics post approval have not changed and the benefit–risk ratio which was used to grant the MA is still favourable. It is obvious that the above efforts result in greater work-sharing leading to a single assessment procedure with a direct reduction in administrative burden. Furthermore, the delivery of a central repository of PSURs will also drive the required regulatory memory to improve consistent decision making across PSUR assessments.
In certain cases after the finalization of the PSUR procedure, a need for further action like the updating of the approved summary of product characteristics may be warranted. Importantly, the outcomes of the assessment are legally binding and on 29 occasions between January 2014 and December 2014, PRAC recommended the update of the product information (www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000375.jsp&mid=WC0b01ac0580727d1c). Another possible outcome of PSUR assessment is revocation of the MA; however, this would be expected to occur only rarely and has not happened in practice [The European Parliament and the Council of the European, 2010a]. This latter point is crucial for MAHs to take note of, as the Directive does not provide any legal mechanism for a reexamination procedure and the opportunity to reverse a recall of the revocation. Thus MAHs should provide solid robust arguments from the beginning of the procedure. The EU PSUR single assessment is commonly referred to as a PSUSA. As a measure to increase risk proportionality and improve efficiency in resources both for MAHs and for NCAs, there is no (routine) requirement for generics, homeopathics, and traditional herbal medicines to submit PSURs. A list of which active substances are not exempt (i.e. that have to submit PSURs) is available on the EU reference dates list (EURD list: www.ema.europa.eu/docs/en_GB/document_library/Other/2012/10/WC500133159.xls). As of November 2014, the EURD list details 3249 active substances (or combinations) with their respective data lock points, as well as identifying 228 active substances requiring the submission of PSURs for generic and well established use medicinal products. Further efficiency gains for MAHs are also felt as a result of the deletion of the requirement that no more (routine) line listings need to be submitted to the regulator. For national authorized medicinal products marketed only in a single EU member state and whose active substance or combination of active substances is included in the EURD list, the MAH should submit a PSUR as part of the PSUSA procedure. The PSUSA involving only NAPs started in the last quarter of 2014 (with data lock points after 1 September 2014). For those national products with active substances or combination of active substances not included in the EURD list, for which a PSUSA procedure has not been established yet, the assessment of PSUR will remain at the national level.
The new pharmacovigilance legislation views member states as a network of assessors and resources with the ultimate objective of safeguarding public health.
Transparency and communication
Major developments have occurred over the last 2 years in the area of communication and transparency. The EMA has launched a website giving public access to EudraVigilance and the ADR reports contained within it, and in 2014 this was expanded to cover substances commonly found in nationally authorized products. The website details alphabetically by product and substance the ADR reports submitted to EudraVigilance. Furthermore, national web portals on safety issues have been set up by national agencies and are linked to the new EU website (http://www.adrreports.eu/en/national.html). Furthermore, NCAs (examples include the UK’s Medicines and Healthcare Products Regulatory Agency, www.mhra.gov.uk; the Malta Medicines Authority, www.medicinesauthority.gov.mt; the Italian Medicines Agency, www.aifa.gov.it; the Irish Health Products Regulatory Authority, www.hpra.ie; Spanish Medicines Agency, www.aemps.gob.es), through their websites, provide not only product information (summary of product characteristics and product information) and safety communications, but also approved Dear Doctor Letters as well as RMMs (for example, patient alert cards, details of registries, educational materials etc.). The NCA websites are host to public assessment reports (including the imposed conditions of MAs), lists of products under additional monitoring and online ADR reporting forms. To date, the EU medicines web portal is served by the EMA website (www.ema.europa.eu), and it is important for industry to follow this as recommendations, conclusions and opinions of the PRAC, CHMP and coordination group posted on the EMA website are now directly applicable to MAHs. Central to all these communication measures is the EMA’s role in coordinating and timing the content of member state safety announcements, including for nationally authorized products outside a formal EU procedure. As a final effort to improve transparency, public hearings linked to pharmacovigilance referrals (to solicit public views and experiences) are being introduced at the level of the PRAC [Pharmacovigilance Risk Assessment Committee, 2014]. It is anticipated that the first such hearings will take place in 2015.
A shift from sending a simple letter to accurately communicate the risk and the warning is observed. Communication towards all directions regulators and agencies and the public is a key element for the new pharmacovigilance legislation. The concept introduced is an effort to educate and train physicians and the public on how to prevent risks. The terms prevention, minimization and proactive measures are crucial for the achievement of the fundamentals of vigilance.
EU database on drug products and active substances
The Article 57 database, referred to in Regulation 1235/2010/EU, is known as the eXtended EudraVigilance Medicinal Product Dictionary (XEVMPD) [The European Parliament and the Council of the European Union, 2010b]. Importantly, MAHs are required to submit information to the XEVMPD for all the products for which they hold a valid MA in the European Economic Area (EEA).
The information submitted to XEVMPD must be maintained and updated by the MAHs, as per the changes normally occurring in the lifecycle of an authorized medicinal product. All other changes have to be performed no later than 30 calendar days from the date when they became effective, as of 1 January 2015 (for variations, 30 calendar days from the approval date). One of the main requirements of XEVMPD is the submission of the summary of product characteristics (SmPC), or in the absence of the SmPC, an equivalent document such as the product information leaflet. This is to allow validation of submitted product information by the EMA. Another requirement is to submit information on all substances contained in the medicinal products, including substance classification (i.e. chemical, specified substance group 1, etc.), bibliographical reference (i.e. company specification, Martindale, etc.) and translations in all languages in which the SmPC exists in the EEA.
According to Article 57 a database will be set up and it will contain all SmPCs and patient leaflets from all medicinal products authorized in the EU, including nationally approved ones. A pool of information after ‘cleaning’ and verification of the data inserted in the database will be created at the end in order to be used by all agencies in EU. This can be considered as a step towards harmonization of the information available of the medicinal products used in member states in Europe and could further enhance vigilance for European citizens, who due to the freedom of travelling can be ‘exposed’ to any medicinal product available in any member state of the EU. Information given in a common format for the same active substance in the same pharmaceutical form will prevent ‘misunderstandings’ in the use of such active substances, including avoidance of misuse or abuse.
For ease of reference, Table 4 lists the new additional fields required. It is important to note that the updates discussed above are, in part, to bridge the future implementation of the International Organization for Standardization Identification of Medicinal Products standards.
New fields added for authorized medicinal products.

Another element that needs to be updated by MAHs is the SME status of the MAH (small, micro, medium enterprise, in which case an SME number must be obtained from the EMA’s SME office and provided in the update, or N/A, for MAHs which do not qualify as SME). This is to allow the calculation of the pharmacovigilance fees.
Inspections
A quite new element introduced with the pharmacovigilance legislation was the mandate to agencies to perform inspections. In order to determine whether MAHs comply with pharmacovigilance obligations established within the EU as well as outside the EU, and to facilitate compliance, NCAs conduct, in cooperation with the EMA, pharmacovigilance inspections of MAHs or any firms assigned to perform MAH pharmacovigilance tasks and activities. NCA inspectors carry out inspections and they will have the power to inspect premises, records, documents, the PSMF of the MAH or any third-party subcontractor contracted by the MAH to fulfil the obligations described in Title IX of Directive 2001/83/EC for pharmacovigilance in accordance with Articles 111(1) and 111(1)(d) [The European Parliament and the Council of the European, 2001]. Accordingly, there is a shift in the area of compliance with an increased reliance on pharmacovigilance inspections (given that the PSMFs replace the current detailed description of the pharmacovigilance system, which is no longer a requirement in the EU). It is important to note that PSMFs must be in place by July 2015 (MAHs should update their dossiers supporting a MA through a Bulk Type IA Immediate Notification (Type IAIN)).
Pharmacovigilance inspections have become mandatory. This should not be interpreted as a ‘policing’ measure, but rather the creation of an environment of direct in person communication and interaction between MAHs and regulators at the site of the MAH and where pharmacovigilance data and reports are kept stored. Inspections can be performed on agencies, including their PSMF systems. The results of such operations can be published, further increasing transparency.
Audit
Article 101(2) of the Directive 2001/83/EC states: ‘Member States shall, perform a regular audit of their Pharmacovigilance system and report the results to the Commission on 21 September 2013 at the latest and then every 2 years thereafter’ [The European Parliament and the Council of the European, 2001]. Similarly, the Regulation imposes requirements for pharmacovigilance audit on the EMA. The CIR requires that NCAs must have an audit system which is risked based [European Commission, 2012] where each NCA’s audit strategy is based on its own risk assessment, taking into account each area that is specified in the CIR. In addition, the NCAs should look into the possibility of auditing areas where nonconformance is not detected by other means (for example, key performance indicators within standard operating procedures) and then understanding the impact such nonconformance could have on public health and the achievement of targets set by the CIR [European Commission, 2012]. Audits carried out by NCAs [European Commission 2012; HALMED, 2013, Medicines Evaluation Board, 2013] and made available in the public domain indicate that NCAs are reviewing areas of activities (see Table 5) and assign a risk rating. According to the audits carried out, the risk assessment is then used to determine the priority areas for audits as well as their frequency [European Commission, 2012; HALMED, 2013; Medicines Evaluation Board, 2013]. The activities in this area indicate that pharmacovigilance audits across the EU are more integrated across the EU since 2012 and that compliance with Directive requirements could be viewed from a ‘collective (NCAs/EMA)’ perspective.
Areas audited and assigned risk level.
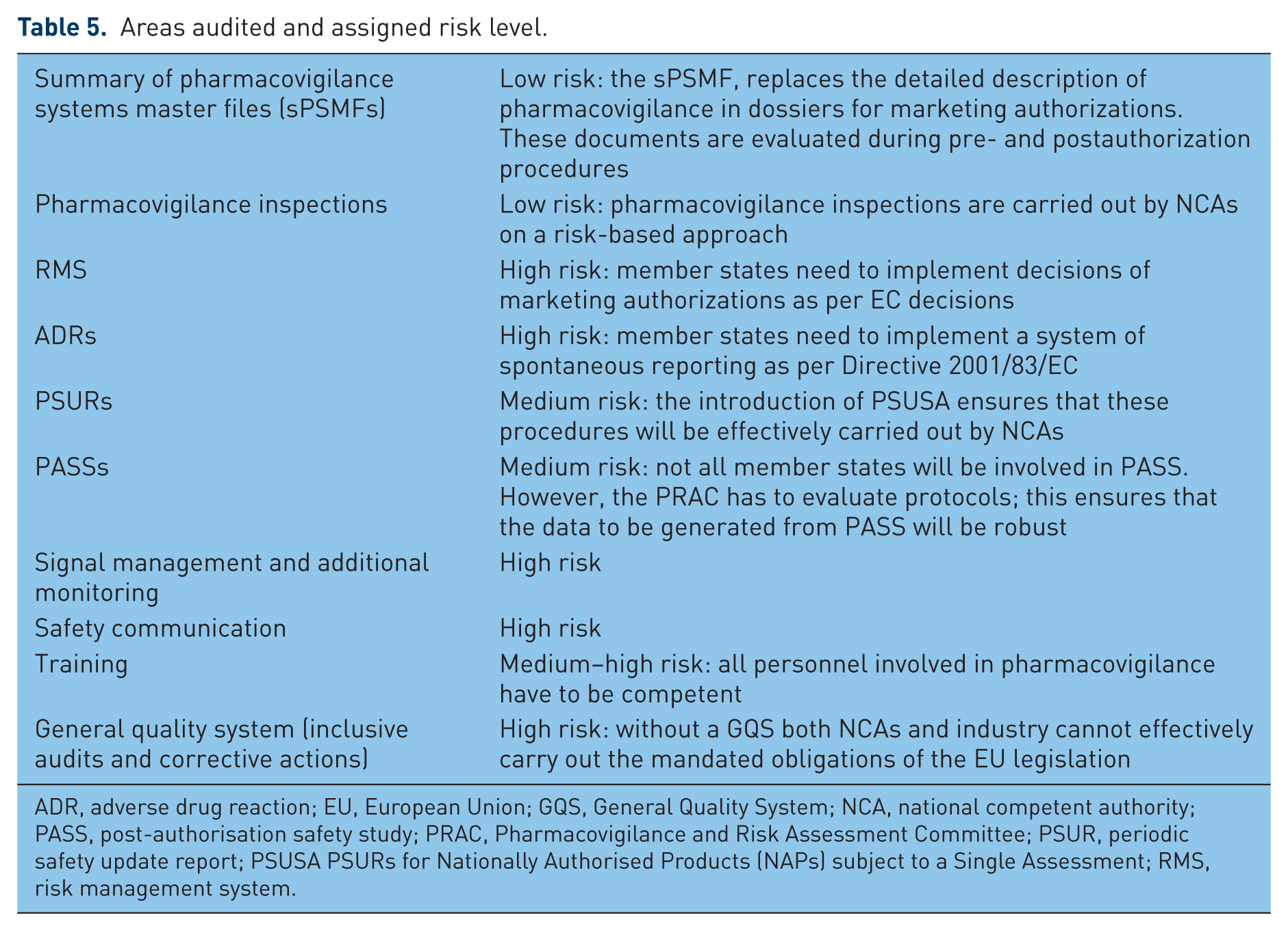
ADR, adverse drug reaction; EU, European Union; GQS, General Quality System; NCA, national competent authority; PASS, post-authorisation safety study; PRAC, Pharmacovigilance and Risk Assessment Committee; PSUR, periodic safety update report; PSUSA PSURs for Nationally Authorised Products (NAPs) subject to a Single Assessment; RMS, risk management system.
Risk management system
A risk management system (RMS) is defined as a set of pharmacovigilance activities and interventions designed to identify, characterize, prevent or minimize risks relating to a medicinal product, including the assessment of the effectiveness of those activities and interventions. A RMP is a detailed description of the RMS for a medicinal product; this requirement was first introduced in the EU legislation (the Regulation) in 2005. For the sake of clarity it is important to note that an RMP summarizes the safety profile of a medicinal product, notes the identified risks, identifies potential risks and lists the further studies that will be carried out post authorization. Any extra RMMs required to be put in place to manage the identified risks of the medicinal product are described in detail. The Directive (Article 106) and Regulation (EC) 1235/2010 (Article 26) specify that a RMP is required for all new applications while the summary of the RMP is also to be made public [The European Parliament and the Council of the European, 2010a, 2010b]. This requirement translates to the following interpretation: for MAs granted after 21 July 2012, MAHs are required to operate a RMS for each medicinal product. Holders of MAs granted before this date are not required to operate a RMS for those medicinal products unless regulators or the MAH are concerned about risks affecting the benefit–risk balance of an authorized medicinal product. Should a RMS for a medicinal product be set up, the MAH is legally obliged to:
(1) monitor the outcome of RMMs which are contained in the RMP or which are laid down as conditions of the MA;
(2) update the RMS and monitor pharmacovigilance data to check for new risks, or to establish whether risks have changed or whether there are changes to the benefit–risk balance of medicinal products.
RMMs are a set of activities which will be done to reduce the risk of an event occurring, or to reduce the harm from the event associated with a particular safety concern. The risks identified for a product are specified in the RMP. There are two types of RMMs:
(1) routine (contraindications and warnings contained within the product information);
(2) additional (activities put in place to reduce the probability of an event occurring through, for example, educational materials for doctors, pharmacists or patients; limiting the size of a package; and having a pregnancy prevention programme, among others. The most commonly reported additional measures used for risk minimization are the educational materials [Zomerdijk et al. 2012].
With respect to the RMP summary submitted for review by applicants in part VI of the RMP, the following elements must be provided.
(1) Overview of disease epidemiology.
(2) Summary of the benefit/efficacy.
(3) Summary of main safety concerns (identified, potential and missing information).
(4) Summary of RMMs by safety concern (routine and additional).
(5) Planned postauthorization (safety and efficacy) development plan.
(6) Major changes over time.
Following the review of the RMP, the summary for the public is shared with the company prior to publication and published at the time and as part of the European Public Assessment Report (EPAR) (at time of Commission decision; this activity started in 2014 at the level of the EMA for CAPs). Importantly, this is linked to the product information, European Public Assessment Report summary and list of medicines under additional monitoring. It is to be kept in mind that the RMP is to be continuously updated during the lifecycle of the medicinal products and updates should be submitted when major changes occur: new indications, restrictions of indication, new/updated contraindications, new important risks or important changes to known risks, and any ‘additional risk minimization measures’ are added or removed. This is within the concept of the continuous circle of collecting data, reporting and evaluating the benefit–risk ratio and confirming whether it remains favourable.
ADR reporting and signal detection
The new legislation defines an ADR as ‘a response to a medicinal product which is noxious and unintended’. Therefore, other causes of ADRs such as MEs, abuse, misuse and occupational exposure are also covered by this definition. Thus member states and MAHs must report ADRs related to MEs to EudraVigilance, which subsequently could be upgraded to be able to better handle these ME reports. This is because different information is required to be captured in an ME report to be able to carry out imputability assessment for MEs compared with causality assessment for adverse events with medicinal products [Tanti et al. 2013]. It is interesting to note that the EU Directive (2001/83/EC) does not codify that near misses of medication errors need to be reported [Tanti et al. 2013]. However, at a national level, regulatory agencies could still be interested in these reports and could introduce national requirements to be able to collect and review near misses (www.medicinesauthority.gov.mt/adrportal). With respect to signal detection, the EU legislation codifies that this activity has to be carried out by the EU network, as well as the MAH, with NCAs leading for NAPs through work sharing and the EMA leading for CAPs. The EMA supports signal detection through the EudraVigilance data warehouse (the EudraVigilance data analysis tool) and electronic reaction monitoring reports which have been developed specifically to carry out signal detection. The EMA defines a safety signal as ‘information on a new or incomplete documented adverse event that is potentially caused by a medicine that warrants further investigation’ [European Medicines Agency, 2013]. In 2013, 2449 potential signals were evaluated, which equates to approximately an 11% increase versus 2012 (2213) and a 54% increase versus 2011 (1586). The signals in 2012 arise from EudraVigilance (91%), the literature (5%), and other rapid alerts (3%). The rapid alert procedure is the procedure used to rapidly communicate among competent authorities of member states the recall of medicinal products if a serious risk to public health has risen with the detection of a defect in a product. The aim of the rapid alert procedure is to transmit only those alerts whose urgency and seriousness for potential harm to patients cannot permit any delay in transmission and to notify all member states of the issue.
Forty-three validated signals by the EMA were further assessed by the PRAC while 69 further potential signals were kept under monitoring at the EMA [European Medicines Agency, 2013].
It should be noted that the increase in signals can be attributed to a great extent to the increased pool of information and the expansion of the EudraVigilance database via the collection of ADRs in the single portal of EudraVigilance and the development of tools for the analysis of these signals.
Conclusion
In this manuscript we have tried to identify the milestones of the EU regulatory network in fulfilling the legal obligations mandated by EU Directives and Regulations. We have attempted to carry out this task by discussing the EU pharmacovigilance network system from a 360° perspective, thus describing the current state of affairs on all the progress made horizontally by EU regulators. The results and data presented show that there has been an intensive effort by regulators and the industry in setting up systems and processes in order to implement the required legislative changes. It is evident that public health decision making has become much more streamlined and transparent with PRAC, which ultimately is being translated into benefits to patients through numerous mechanisms; noteworthy are harmonized safety labelling changes, regulatory action and educational materials for prescribers and when necessary for patients. However, there are comments in the public domain that indicate that the administrative burden with the new system has not decreased compared with pre 2012 [Daniels, 2013; Schofield 2014] and it is noted that during 2015 and 2016 new services and systems will come online to reduce the burden on the industry (for example, the EMA literature monitoring service). In conclusion, the experience gained so far with changing trends of discussion topics every month at the PRAC indicate that it is still too early to arrive at an informed decision as to whether the legislative tools provided by the Directive are being utilized in a maximal manner. However, from the data presented, we observe that within the EU, a major improvement in implementing safety decisions through a rationalized and harmonized approach is occurring. This must surely be viewed as a major public health contribution of the new EU legislative framework.
Footnotes
Acknowledgements
The authors wish to thank Dr Peter Arlett for his comments during the drafting of this manuscript. The views expressed in this article are the personal views of the authors and may not be used or quoted as being made on behalf of, or reflecting the position of, any national competent authority, the EMA or one of its committees or working parties or any University.
Conflict of interest statement
The author declares no conflicts of interest in preparing this article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.