
Abstract
Almost three decades after the publication of the first clinical studies with tacrine, the pharmacological treatment of Alzheimer’s disease (AD) remains a challenge. Randomized clinical trials have yielded evidence of significant – although modest and transient – benefit from cholinergic replacement therapy for people diagnosed with AD, and disease modification with antidementia compounds is still an urgent, unmet need. The natural history of AD is very long, and its pharmacological treatment must acknowledge different needs according to the stage of the disease process. Cognitive and functional deterioration evolves gradually since the onset of clinical symptoms, which may be preceded by several years or perhaps decades of silent, presymptomatic neurodegeneration. Therefore, the pharmacological treatment of AD must ideally comprise both a symptomatic effect to preserve or improve cognition and a disease-modifying effect to tackle the progression of the pathological process. Primary prevention is the ultimate goal, should these strategies be delivered to patients with preclinical AD. In this article, we briefly address the pharmaceutical compounds that are currently used for the symptomatic treatment of AD and discuss the ongoing strategies designed to modify its natural course.
Introduction
In the past two decades, the body of knowledge on pathophysiological mechanisms of Alzheimer’s disease (AD) has increased substantially. Pathological cascades including extracellular amyloid plaques, intracellular neurofibrillary tangles, inflammatory mechanisms, synaptic degeneration and loss of neuronal cells have been extensively investigated [Braak and Del Tredici, 2011; Winblad et al. 2014; Sperling et al. 2014]. Convergent findings strongly suggest that these neurobiological mechanisms start long before clinical manifestations, although it is debatable how, where and when the neurodegenerative process actually begins [Dubois et al. 2014; Sperling et al. 2014; Jack et al. 2014]. A better understanding of pathological mechanisms of the disease allows the proposal of new effective and safe treatment strategies.
Currently, there are three acetylcholinesterase (AChE) inhibitors – donepezil, galantamine and rivastigmine – available for AD treatment, as well as the glutamatergic antagonist memantine. Despite being widely prescribed, these drugs have only limited and transient symptomatic effects when compared with placebo, without a definite impact on the long-term neurodegenerative process [O’Brien and Burns, 2011].
Other pharmaceutical compounds were designed to attenuate or interrupt the progression of neurotoxic events subsequent to overproduction of the amyloid-beta peptide (Aβ), with potential effects in animal models. However, clinical trials with most of these agents have not so far provided any conclusive evidence of clinical benefit for human patients, in spite of promoting Aβ clearance and/or attenuation of neuropathological changes within the brain.
Converging efforts toward the development of pharmacological agents with proprieties to effectively prevent or interrupt the earliest stages of the neurodegenerative downstream pathways are in still progress and remain a worldwide crucial issue [Sperling et al. 2014; Kivipelto et al. 2014]. Likewise, disease-modifying compounds, which inhibit extra-neuronal Aβ deposition or interrupt intraneuronal tangles formation (NFT), at the same time providing favorable clinical efficacy, constitute an urgent expectation for treatment [Winblad et al. 2014]. In addition, to recognize the main predictors for conversion from non dementia to dementia condition and to control risk factors may give desirable selectivity in achieving preventive strategies.
This review aims to discuss three main aspects focusing on: (a) currently available therapy for symptomatic treatment of patients with AD, which have been driven to preserve or improve cognition and functionality; (b) new ongoing pharmaceutical compounds originally designed to change the disease course by blocking or slowing neurodegenerative downstream related to Aβ peptide and tau protein; and (c) primary prevention based on nonpharmacological approaches involving lifestyle and supportive interventions with delaying the disease onset as the main purpose.
Symptomatic pharmacological agents
Pharmacological mechanisms of AChE inhibitors are related to a diminished cholinergic synaptic activity due to reduction of choline acetyltransferase, an enzyme directly responsible for acetylcholine synthesis in the basal nucleus of Meynert [Perry et al. 1977].
Agents as donepezil, galantamine and rivastigmine inhibit the catabolic enzyme AChE to maintain or at least to delay the decrease of acetylcholine levels in synaptic clefts and are recommended for patients with mild to moderate AD. Despite being widely prescribed, the magnitude of clinical efficacy of the three compounds remains modest and symptomatic, and usually for a limited period, without evidence of substantial impact on disease course [Di Santo et al. 2013; Gauthier et al. 2013 Amanatkar and Grossberg, 2014]. All three drugs share the anticholinesterasic property and two of them act through distinct pharmacodynamic mechanisms.
Donepezil is a highly selective and reversible central AChE inhibitor at standard doses of 5–10 mg per day. Patients at moderate to severe AD could benefit from a higher dose of 23 mg per day; this dosage conferred a slight efficacy over 10 mg, with worsening of side effects such as gastrointestinal complaints during the first month of treatment, but with decreased occurrences thereafter [Christensen, 2012].
Rivastigmine inhibits both AChE and butyrylcholinesterase. Prescriptions start with 1.5 mg and should be progressively increased to 6 mg twice a day. The transdermal patch has a long halflife, allowing prescription once a day at higher dosages with lower gastrointestinal side effects, and also for patients at more severe stages of the disease, when recommendable [Amanatkar and Grossberg, 2014].
With specific pharmacodynamics properties, galantamine is a reversible and competitive inhibitor of AChE, and is also an allosteric modulator by binding to cholinergic nicotinic receptors at an additional site [Grossberg, 2003; Devanand, 2014]. A daily extended-release prescription ranges from 16 to 24 mg.
The most common side effects of AChE inhibitors are gastrointestinal manifestations such as nausea, vomiting, diarrhea and weight loss; insomnia, headache, agitation, bradycardia and syncope are less common. With a careful titration over 3 months, the tolerability of the AChE inhibitors seems to be similar, without clinically significant differences among them [Birks, 2006].
The neurodegenerative process of AD comprises several neurotoxic mechanisms including abnormalities of excitatory amino acids such as glutamate, the most widespread excitatory neurotransmitter. Its excitotoxicity is closely linked to chronic calcium (Ca++) influx through Ca++ channels of N-methyl-
Memantine is an uncompetitive glutamatergic receptor antagonist that engenders a blockage or regulation of NMDA receptors, providing a more suitable neurotransmission required for synapses functioning [Winblad et al. 2010]. Although the combination of AChE inhibitors with memantine seems to be safe and rational (because of distinct mechanisms of action), a study reported no conclusive clinical evidence to recommend this strategy for AD treatment [Gauthier et al. 2012]. Another investigation evaluated the efficacy, safety and tolerability of the extended released-memantine formulation with higher dose (28 mg once daily) in outpatients with moderate-to-severe AD including patients on stable AChE inhibitor [Grossberg et al. 2013]. The authors reported a significant advantage of this formulation over placebo regarding clinical measures. Nonetheless, this study did not directly compare the extended-release formulation of memantine (28 mg once daily) with standard dosing.
Disease-modifying therapies
Anti-amyloid-β passive and active compounds
Although intensely debatable, the amyloid cascade hypothesis of AD pathology assumes that deregulation in amyloid precursor protein (APP) leads to accumulation of Aβ1–42 peptide in the brain parenchyma; in addition to this hypothesis, other related-mechanisms such as synapsis loss, tau phosphorylation, NFT formation and neuronal degeneration have been established [Winblad et al. 2014; Sperling et al. 2014; Jack et al. 2014].
Cleavage mechanisms of APP implicate two and mutually exclusive pathways: the nonamyloidogenic (or secretory pathway) and the amyloidogenic pathway. In the nonamyloidogenic pathway, APP is first cleaved by α-secretase, releasing a soluble N-terminal (sAPPα) and a C-terminal fragment, which is cleaved by γ-secretase to originate a smaller C-terminal fragment [Roberts et al. 1994; Recuero et al. 2004; Devanand, 2014].
In the amyloidogenic pathway, APP is alternatively cleaved by β-secretase releasing a smaller N-terminal fragment (sAPPβ) and a longer C-terminal fragment that contains the full amyloidogenic sequence of amino acids. A further cleavage of APP by γ-secretase yields the Aβ peptide. Aβ1-42 is prone to aggregation, being the most neurotoxic Aβ peptide in the AD pathogenesis [Recuero et al. 2004]. Aβ oligomers are considered the most toxic forms of the amyloid derivates [Roberts et al. 1994; Devanand, 2014]. They interact with neurons and glial cells leading to several pivotal mechanisms that ultimately lead to cell death.
Over the past years, passive and active immunotherapies have been developed whose the main purpose is to change the course of AD pathology through the efflux of Aβ deposits from the brain to the peripheral circulation, or the reduction of parenchymal Aβ peptide aggregation.
With respect to passive immunization, solanezumab, bapineuzumab, gantenerumab, crenezumab and ponezumab are currently being tested with the purpose of inducing the clearance of brain Aβ burden and to improve clinical outcomes with the expectation of favorable impact on cognition and functionality. Preliminary results from randomized clinical trials involving different compounds have achieved these goals with acceptable safety, while others brought disappointing neurobiological and clinical results or were related to serious adverse occurrences (Table 1).
Current pharmacological interventions for Alzheimer’s disease: acetylcholinesterase inhibitors, anti-amyloid-β drugs, anti-tau compounds.
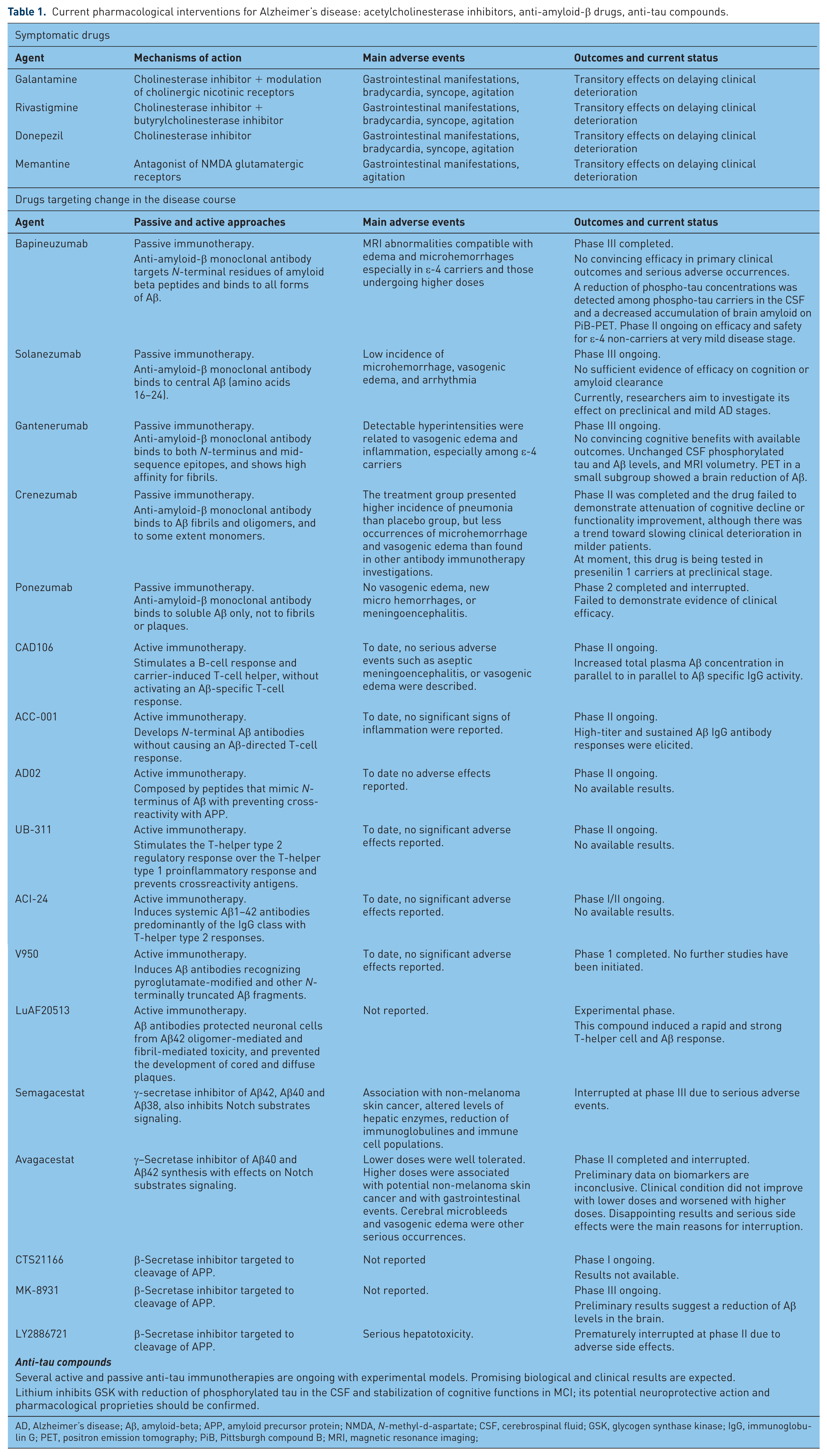
AD, Alzheimer’s disease; Aβ, amyloid-beta; APP, amyloid precursor protein; NMDA, N-methyl-
Bapineuzumab is a humanized anti-amyloid-β monoclonal antibody intravenously administered to patients with mild-to-moderate AD currently at phase III clinical trial. Unfortunately, this agent did not demonstrate convincing efficacy on cognition or basic daily living activities. In general, neither carriers nor noncarriers of the ε-4 allele of apolipoprotein E presented significant benefits with bapineuzumab. Interestingly, ε-4 carriers exhibited reduction of phospho-tau concentrations in the cerebrospinal fluid and decrease of amyloid accumulation on Pittsburgh compound B (PiB-PET) studies [Salloway et al. 2014]. Magnetic resonance imaging (MRI) abnormalities suggestive of vasogenic edema and sulcal effusions (ARIA-E; incidence of 17.1%) and hemosiderin deposits (ARAIA-H; incidence of 9.2%) compatible with microhemorrhages were reported in 17% of treated patients, being more frequent among ε-4 carriers and those in ascending dose regimen [Sperling et al. 2012]. The phase III clinical trial with bapineuzumab was interrupted due to disappointing clinical outcomes and serious side effects. Subsequently, researchers started a new arm from previous investigation, currently at phase II, to test potential efficacy and safety of earlier treatment with bapineuzumab for a pooling of ε-4 noncarriers patients at very mild disease stage [Moreth et al. 2013].
At phase III trial, the humanized monoclonal antibody solanezumab intravenously administered failed to achieve favorable results. Despite a subsample comprising patients with mild dementia where it seemed to present some cognitive or functional benefits, specific measurements did not provide convincing evidence of clinical improvement [Doody et al. 2014]. Furthermore, this drug had no efficacy in promoting clearance of Aβ from brain parenchyma of mild-to-moderate AD patients, although the authors reported inconclusive results on accumulation of this protein according to positron emission tomography (PET) with F-florbetapir tracer [Doody et al. 2014]. Likewise, this treatment was not effective in slowing the atrophy of whole brain or hippocampal volumes assessed by structural MRI. Although the authors reported low occurrences of side effects, patients treated with solanezumab presented adverse events as microhemorrhage, vasogenic edema, arrhythmia, and skin and subcutaneous tissue disorders, among others, although without statistically differences compared with the placebo group. In order to test the efficacy of this agent in selected groups, the authors suggested the development of further studies in patients at a mild stage of AD pathology or in individuals at preclinical phase who exhibit evidence of brain amyloid accumulation [Doody et al. 2014]. Another upcoming trial was designed to determine if solanezumab potentially prevents the disease development in large families with genetic mutations for AD pathology [Moreth et al. 2013].
Another randomized controlled trial was designed to test the efficacy and safety of gantenerumab in patients with mild AD [Ostrowitzki et al. 2012; Moreth et al. 2013; Panza et al. 2014]. Currently at phase III, this agent is another humanized anti-amyloid-β monoclonal antibody subcutaneously administered, targeting both terminal and central regions of fibrillar Aβ [Panza et al. 2014]. Primary outcomes related to cognition and functionality were not significantly favorable over placebo. Secondary outcomes concerning total tau, phosphorylated tau and Aβ levels in cerebrospinal fluid, as well as MRI volumetry from patients treated with gantenerumab, were similar to those found in controls. Conversely, a PET study in a small subgroup showed a reduction of Aβ deposition in the brain. Detectable FLAIR hyperintensities were ascribed to excessive pharmacological activity consistent with vasogenic edema and inflammation, especially among ε-4 carriers [Ostrowitzki et al. 2012; Moreth et al. 2013]. At this time, gantenerumab is being tested as a potential preclusive of the disease progression in individuals with genetic mutations for the AD pathology [Moreth et al. 2013].
Crenezumab is also a humanized monoclonal antibody, administered by intravenous or subcutaneous injections, addressing to clean all forms of Aβ peptide from brain parenchyma in mild to moderate AD [Adolfsson et al. 2012; Mikulca et al. 2014]. Expected mechanisms of action of this drug involve binding to both soluble monomeric and oligomeric forms of Aβ, and subsequently the aim is to prevent its aggregation or to dissolve plaques already established. A phase II trial was completed in which crenezumab unfortunately failed to demonstrate attenuation of cognitive or functional decline [Mikulca et al. 2014; Cummings, 2014]. Furthermore, preliminary results from a phase II trial involving asymptomatic individuals with autosomal dominant mutations for AD pathology were not significant [Panza et al. 2014]. Currently, this agent is being tested in presenilin 1 carriers at preclinical stage of the disease. With respect to safety, patients receiving crenezumab presented higher (3.2%) incidence of pneumonia than the placebo group (0.6%); however, they exhibited less occurrences of microhemorrhage and vasogenic edema when compared with other antibody immunotherapy investigations [Adolfsson et al. 2012; Cummings, 2014].
Ponezumab, another humanized anti-amyloid-β monoclonal antibody intravenously administered, demonstrated no clinical efficacy at phase II and has been discontinued, in spite of some results suggesting that a soluble pool of Aβ in the brain might be changed with treatment, in absence of peripheral immune response [Landen et al. 2013]. The authors reported absence of vasogenic edema, new microhemorrhages or meningoencephalitis.
Other passive immunotherapeutic approaches toward changing the Aβ cascade are ongoing in clinical trials. Currently at phase I or II, some compounds are being investigated and results have not been published yet: BAN2401, GSK933776, AAB-003, SAR228810 and BIIB037/BART [Winblad et al. 2014].
Strongly encouraging, neurobiological data from the first generation of active immunotherapy were tested in experimental models, reducing amyloid burden from brain parenchyma and providing the rationale for subsequent investigations in AD patients. In this context, AN1792 was the first active Aβ immunotherapy in clinical trials with patients at mild-to-moderate AD disease stages. Although this compound was effective in reducing Aβ from brain parenchyma and in providing some cognitive benefits, the trial was interrupted at phase II because only 19.7% of patients presented antibody response to this treatment, and 6% developed serious cytotoxic T-cell reactions and acute meningoencephalitis [Gilman et al. 2005].
Unlike previous trials, those addressing active Aβ immunotherapies from the second generation have focused on efficacy and safety, and included early AD stages. In this way, trials with CAD106, ACC-001, AD02, ACI-24, V950, UB-311 and LuAF20513 compounds were designed to avoid inflammatory reactions, microhemorrhage or vasogenic edema, and are currently at phases I or II with promising preliminary results [Winblad et al. 2014].
γ-Secretase and β-secretase inhibition
Approaches comprising investigations based on other disease-modifying strategies are ongoing. For instance, there is a growing knowledge body on compounds involving inhibition of γ-secretase and β-secretase in order to preclude amyloidogenic cleavage of APP.
The inhibitor of γ-secretase, semagacestat, orally administered, was developed to potentially reduce the Aβ downstream pathway and was tested in patients with mild-to-moderate AD. However, this trial was interrupted at phase III because this compound was associated with serious adverse effects such as nonmelanoma skin cancer, altered levels of hepatic enzymes, reduction of immunoglobulines and immuno-cell populations [Doody et al. 2013]. Although the authors have reported a decreased level of phospho-tau in a subsample receiving high doses of semagacestat when compared with placebo group, cerebrospinal levels of Aβ were unchanged and clinical results were disappointing. Actually, cognitive and functional measurements worsened among all groups. Moreover, there were no significant differences on fluorodeoxyglucose (FDG) PET and F-florbetapir-PET measures among treatment and placebo groups.
Avagacestat, an oral administration compound, is also a γ-secretase inhibitor designed for selective inhibition of Aβ in patients with prodromal AD, currently on phase II trial [Istace et al. 2014]. Data related to cerebrospinal fluid biomarkers, such as Aβ peptides and phospho-tau, were inconclusive. Controversial outcomes were reported also for clinical measurements. Cognitive and functional performance from patients using lower doses did not substantially change and performance from those undergoing higher doses were worsened [Coric et al. 2012]. As reported, lower doses were well tolerated, whereas higher doses of avagacestat were associated with nonadmissible rates for continuation, which were attributed to potential nonmelanoma skin cancer, cerebral microbleeds, vasogenic edema, and gastrointestinal events such as diarrhea and vomiting [Coric et al. 2012]. Paradoxically, avagacestat-treated patients exhibited greater cerebral volume loss compared with the placebo group, suggesting brain amyloid lowering [Istace et al. 2014], even though this hypothesis was not confirmed. Disappointing clinical and neurobiological results, as well as potential serious adverse effects were the main reasons for discontinuation of this trial [Mikulka et al. 2014].
There are several agents addressing the inhibition of β-site APP-cleaving enzyme 1 (BACE-1), a β-secretase responsible for cleavage of APP into Aβ40 and Aβ42 sites. Some trials involve compounds at phase I (CTS21166) and phase III (MK-8931) for prodromal and mild AD, while another (LY2886721) was prematurely interrupted due to hepatotoxicity [Davis and Couch, 2014]. According to preliminary data, the inhibition of β-secretase through MK-8931 had no efficacy in promoting Aβ clearance from brain parenchyma [Menting and Claassen, 2014] (Figure 1).
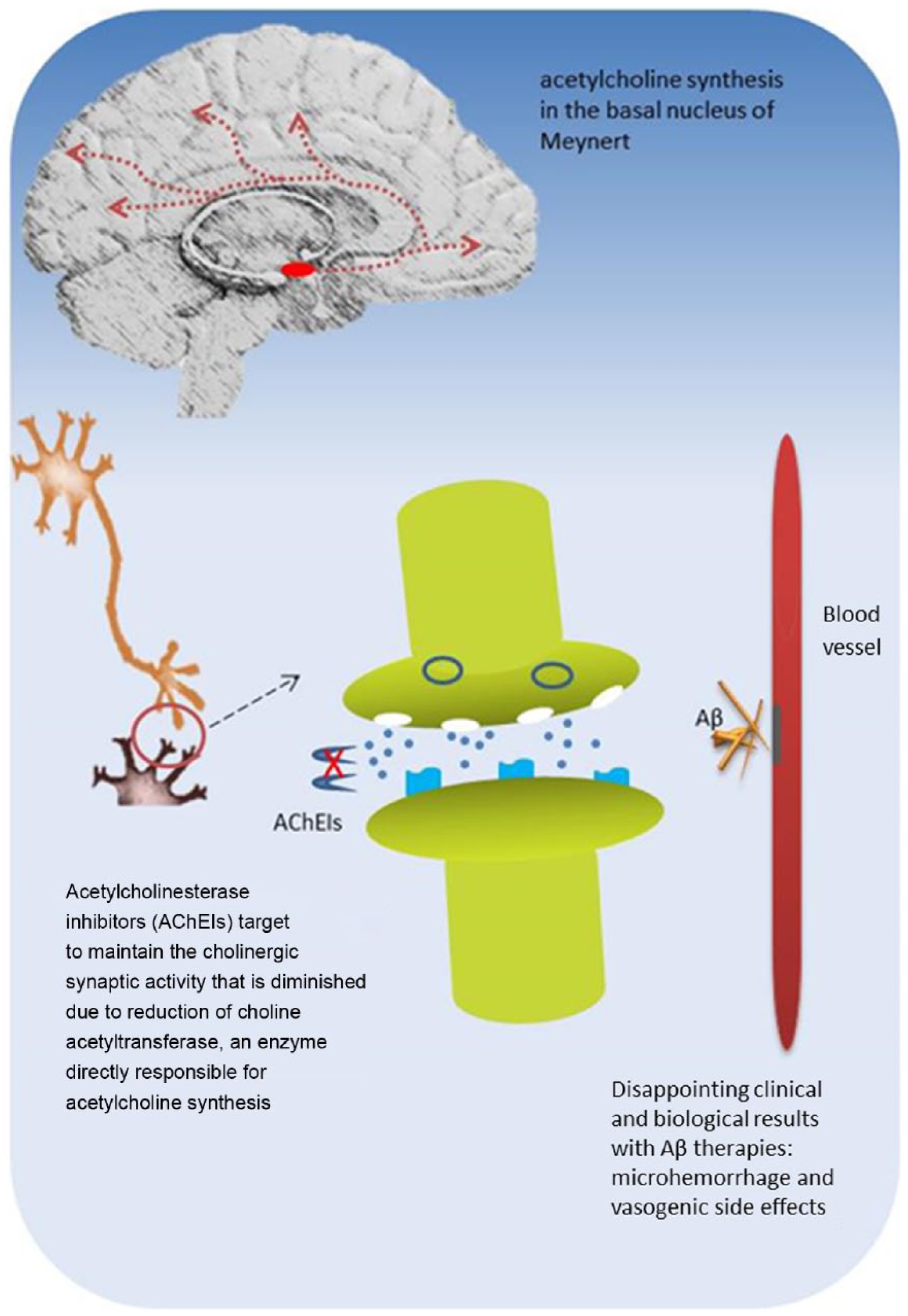
Illustrative depiction of acetylcholinesterase inhibitors and drugs related to amyloid-β mechanisms in Alzheimer’s disease.
Tau-related immunotherapy
In general, studies have given limited information on the onset or later clearance of NFT, which are the most prominent and mature form of tau pathology to appear in the brain from patients with distinct neurodegenerative illnesses, including AD. Tau protein may be found in soluble and insoluble forms, the latter being identified in paired helical filaments (PHF), which are the main component of NFT. PHF-tau complexes have six to eight phosphate groups per molecule of tau protein, bringing much higher than the usual degree of tau phosphorylation observed in the healthy brain (i.e., two phosphate groups per molecule)
Since recent clinical trials that sought to reduce abnormal amyloidogenic process have not provided convincing results, notably on cognitive functions, other approaches are coming with distinct methodological designs. It is strongly accepted that NFT, synapses loss and neuronal death are associated with clinical deterioration in AD. So, active and passive tau-directed immunotherapies involving experimental models are in progress with promising results. Reducing the levels of tau oligomers, preventing tau aggregation and blocking hyperphosphorylation or microtubule destabilization could preclude the neurofibrillary pathologies and constitute thorough aims from new ongoing immunotherapy studies [Herrmann and Spires-Jones, 2015]. One example regards to the experimental investigation with the active vaccine DC8E8 in transgenic mice aiming to prevent tau aggregation and to reduce the amount of a wide range of tau oligomers and neurofibrillary tangles in the brain [Kontsekova et al. 2014].
Despite the promising avenue through anti-tau immunotherapy, one technical problem concerns the discrimination between physiological and pathological mechanisms within tau protein; obviously, the proposed design for anti-tau compounds should be targeted exclusively to inhibit mis-disordered species.
As regard other agents with disease-modifying properties, lithium may prevent tau phosphorylation through glycogen synthase kinase 3 beta (GSK3β) inhibition. Epidemiological investigations demonstrated a reduced risk of progressing to dementia among bipolar disorder patients in long-term treatment with lithium [Nunes et al. 2007]. In addition, patients with amnestic mild cognitive impairment (MCI) showed cognitive and functional stabilization with long-term treatment with lithium, also with reduced phosphorylated tau in cerebrospinal fluid, making this therapy a promising strategy [Forlenza et al. 2011]. Table 1 presents a summary of current pharmacological agents in AD.
Studies with other compounds with potential benefits are in progress, but results are not yet conclusive. A meta-analysis to evaluate the efficacy of ginkgo biloba extract 761, at daily doses of 240 mg, reported benefits for patients with dementia, with acceptable tolerability [Gauthier and Schlaefke, 2014]. However, a review comprising several pharmacological compounds found positive and negative results with ginkgo biloba, as well as negative outcomes with oral anti-inflammatory agents, statins and omega-3 fatty acids; this review also reported controversial results with high doses of vitamin E (2000 IU daily?) [Devanand, 2014]. A meta-analysis addressing dietary intakes of vitamin E, vitamin C and β-carotene suggested a protective effect of each compound against the risk of progression to AD [Li et al. 2012]. Moreover, a randomized and placebo controlled trial with vitamin B treatment (folic acid, vitamin B6 and vitamin B12) enrolled elderly people with MCI over a 2 year follow-up period [Douaud et al. 2013]. The authors demonstrated that high doses of vitamin B reduced gray matter atrophy in regions particularly vulnerable to neurodegenerative processes related to AD such as the hippocampus, parahippocampal gyrus, parietal lobe and retrosplenial cortex. They attributed the atrophy reduction to the lowering of mean plasmatic levels of homocysteine.
The combination of memantine plus molecules having antioxidant effects, as vitamin D, may provide additional clinical benefits for treatment of AD. Currently, a new compound [ClinicalTrials.gov identifier: NCT01409694] is being investigated with the hypothesis of increasing neuronal protection against the disease progression [Annweiler and Beauchet, 2012].
Some drugs with distinct mechanisms of action including anti-inflammatory proprieties and potential interference in Aβ processes have been reported as promising candidates for further treatment of patients with AD, for instance, curcumin, apigenin and tenilsetam [Millington et al. 2014]. A study has demonstrated that a ginseng derived-compound attenuated amyloid deposition in the brain parenchyma of mice models, although the molecular mechanisms of exerted effects are not completely elucidated [Hwang et al. 2012]. Apigenin (4′,5,7-trihydroxyflavone) is a pharmacologically active compound currently at the experimental stage. Molecular tests in mice have demonstrated a lowered insoluble Aβ burden, suppression of amyloidogenic process and inhibition of oxidative stress, with improved learning [Zhao et al. 2013]. Since AD is associated with a chronic inflammatory response to neurodegenerative processes, anti-inflammatory drugs such as tenilsetam may attenuate proinflammatory mechanisms with potential benefits for cognitive functions in AD [Millington et al. 2014]. Some decades ago, a pilot trial over a 3-month period showed significant cognitive improvements particularly in judgment task and reaction time in AD patients [Ihl e al., 1989].
Curcumin is an anti-inflammatory and antioxidant compound with Aβ binding properties. Recently, a study reported that an inhalable formulation was effective to delivery curcumin to critical brain regions related to AD pathology as the hippocampus in mice models [McClure et al. 2015]. Conclusive results from experimental procedures supporting potential benefits for human beings are required.
Most investigations on new drugs are being held with patients with mild-to-moderate disease. Efficacy, tolerability and potential adverse effects, as well as the co-occurrence of medical illnesses in patients with advanced AD, remain questions to be explored.
Interventions at the preclinical stage of AD
In general, when clinical trials begin to investigate patients with mild or moderate AD, the pathological cascade of the disease had already started many years before with synapsis deterioration, neuronal death and widespread disorganization of neurochemical and physiological systems, leading to subsequent brain atrophy and clinical deterioration. Disappointing outcomes at least in part could be attributed to an advanced pathological stage of the disease when the patients were enrolled for trials even at the mild phase of dementia.
In collaboration with the Alzheimer’s Prevention Initiative (API), crenezumab is currently being tested in a large Colombian cohort of patients at an early phase of the disease, also as a potential preventive intervention in presenilin 1 carriers [Reiman et al. 2014; Sperling et al. 2014].
Currently, solanezumab and gantenerumab are being tested in individuals at high risk for AD by an international consortium (The Dominant Inherited Alzheimer Network - DIAN). At phase II, these trials seek to estimate the potential effects of these agents in asymptomatic to mildly symptomatic presenilin 1 and presenilin 2 carriers, as well as in those with Aβ precursor protein (APP) mutations [Mills et al. 2013; Sperling et al. 2014].
Furthermore, international collaboration programs have brought together researchers from different centers with the purpose of developing strategies for primary prevention of dementia in large samples, involving lifestyle changes and long-term control of cerebrovascular risk factors. Programs as the International Database on Aging and Dementia (IDAD), the European Dementia Preventive Initiative (EDPI) and the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), among others, are in progress and represent an important window for primary preventive strategies aimed at people at risk of dementia.
One relevant matter concerns the methodological design of trials comprising Aβ antibody alone. Since Aβ plaques and neurofibrillary tangles are the hallmarks of AD pathology, and considering the strong interface between them, interventions combining both mechanisms may have a synergistic action. So, therapeutic approaches designed to embrace both Aβ and tau phosphorylation in patients at preclinical stages of the disease may have more efficacy than targeting one compound alone, especially when the pathology is potentially reversible [Laske, 2014]. Likewise, integrating approaches covering distinct pathological ways may constitute a promising avenue for new disease-modifying agents [Lambracht-Washington and Rosenberg, 2013].
Nonpharmacological support and prevention approaches
Efforts to develop alternative symptomatic and supportive interventions have been encouraged since potential disease-modifying treatments are not yet available. In this scenario, general healthcare, preventive programs, brain stimulation and cognitive reserve promotion remain crucial to preserve normal functionality.
Adjuvant nonpharmacological resources could eventually be recommended (Table 2). Recently, three medical foods – axona, cerefolin NAC and souvenaid – have been available. So, by supplying ketone bodies as an alternative energy to neurons, axona represents an example of this nonpharmacological intervention. Cerofolin NAC has a favorable role on oxidative stress related to memory loss. Souvenaid provides a nutrient combination thought to enhance synaptic function, maintain integrity of the neuronal membrane, increase resistance to oxidative stress and preserve metabolic activity in the brain [Kamphuis et al. 2011].
Non-pharmacological interventions.

Lifestyle changes comprising long-term control of modifiable risk factors for cerebrovascular disease, as well as a healthy diet, smoking avoidance and regular aerobic physical exercise may contribute to delaying the beginning of clinical deterioration of asymptomatic patients with AD pathology [Sofi et al. 2011; Sperling et al. 2014] (Table 2).
In addition, higher cognitive reserve built through long-term engagement in complex cognitive activities may enhance synaptogenesis and neuronal interconnections that are related to brain reserve, potentially reducing the risk for Aβ downstream cascade or postponing clinical deterioration [Landau et al. 2012; Sperling et al. 2014]. Figure 2 shows a general scheme involving preclinical and clinical interventions in AD, as well as brain and cognitive reserve as protective actions.
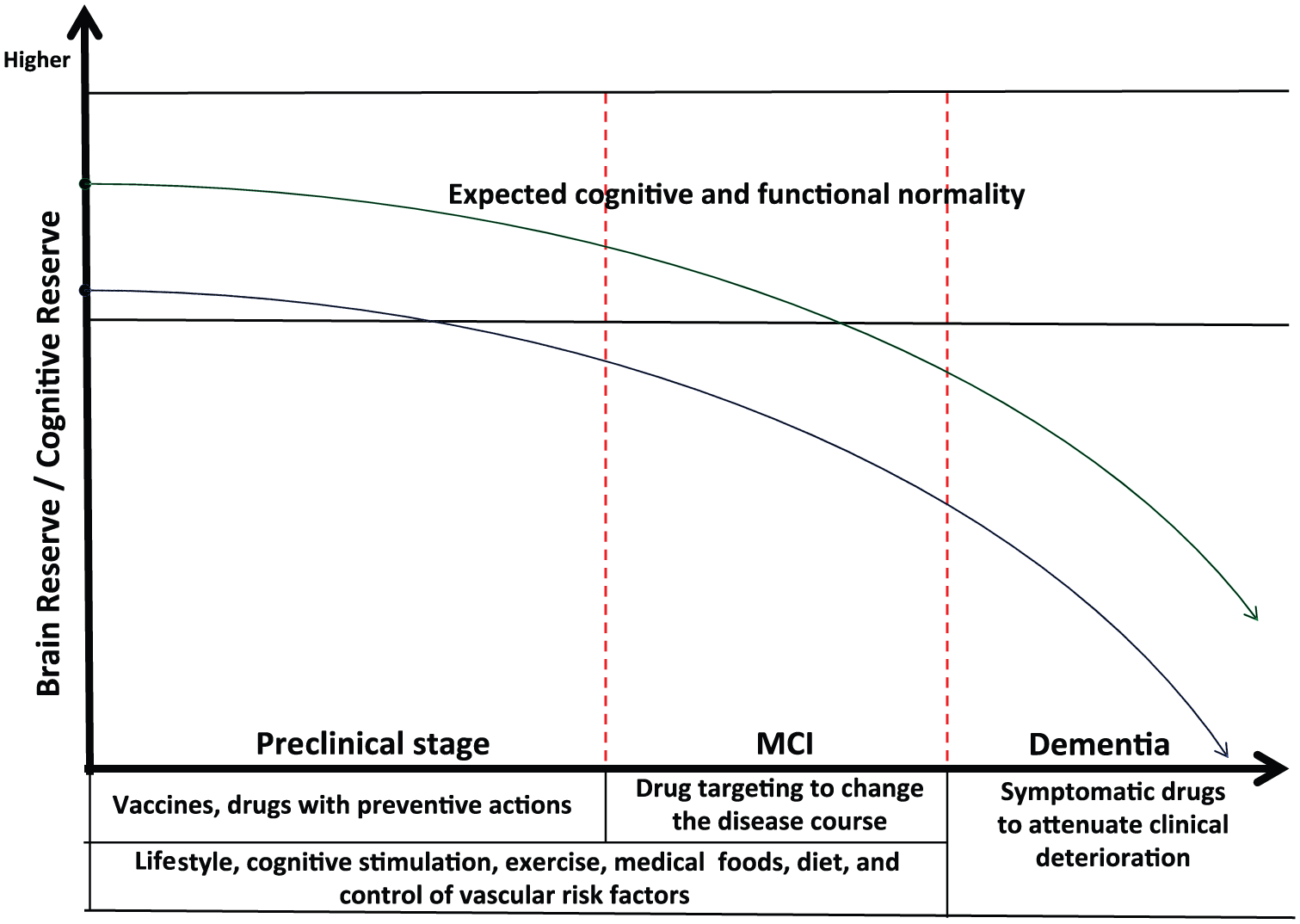
General scheme involving preclinical and clinical interventions in Alzheimer’s disease. Higher cognitive and brain reserve (green line) may attenuate clinical deterioration in comparison with lower reserve (blue line).
Conclusion
Some investigations are ongoing with expectation to change the course of AD, although others have been interrupted due to serious side effects or failed to demonstrate biological or clinical efficacy. In general, therapies have been unable to change the disease course as they are by definition instituted too late, when patients are already strongly affected by the pathological mechanisms. Currently, a growing body of knowledge allows more appropriate methodological designs and enrollment of highly selected patients. Hence, nowadays there is a strong tendency to investigate patients even at preclinical stages, or individuals at risk of the disease, aiming to interrupt neurodegenerative processes. As Aβ and tau phosphorylation seem to be convergent pathological mechanisms, a combination therapy covering both processes could represent a breakthrough for AD treatment. Moreover, in this scenario, preventive interventions based on selected biological agents with nonpharmacological approaches should be encouraged.
Footnotes
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.
Funding
Funding was provided by Fundação de Amparo à Pesquisa de São Paulo (FAPESP Grant nº 09/52825-8, Brazil) and Associação Beneficente Alzira Denise Hertzog da Silva (ABADHS).