
Abstract
Background:
β-Thalassemia major patients are frequently vulnerable to endocrine dysfunction due to iron overload from chronic transfusions. This impairs growth, thyroid function, glucose metabolism, and bone health, ultimately compromising quality of life and long-term outcomes.
Objectives:
This study investigates the prevalence and pattern of endocrine dysfunction in β-thalassemia major patients receiving iron-chelation therapy and explores associations with iron overload markers.
Methods:
This case-control study involved 60 β-thalassemia major patients and 20 age- and sex-matched controls. Hormonal and biochemical parameters were measured and linked to iron status.
Results:
Among β-thalassemia major patients (7–35 years), 73.3% (n = 44/60) were splenectomized; 36 received deferiprone, 19 deferasirox, and 5 deferoxamine. Concerning iron status, both splenectomized and non-splenectomized patients had significantly higher iron and ferritin and lower haptoglobin levels compared to controls. No significant differences were found in hepcidin or hemopexin levels. Regarding thyroid function, about 15% (n = 9/60) of β-thalassemia major patients had subclinical primary hypothyroidism. Ferritin negatively correlated with free thyroxine (r = −0.330, p = 0.010). As for glycemic status, 51.7% (n = 31/60) of β-thalassemia patients had glycated hemoglobin (HbA1c) ⩾6.5% and 38.3% (n = 23/60) showed impaired fasting blood sugar. With respect to metabolic markers, splenectomized patients had higher fibroblast growth factor 21 (FGF21) than the control (p = 0.042), while no significant group differences were found in galectin-1 or sortilin. Ferritin correlated significantly and positively with FGF21 levels (r = 0.353, p = 0.006). With respect to calcium–parathyroid–vitamin D axis, hypoparathyroidism and hyperparathyroidism were each found in 11.7% (n = 7/60) of β-thalassemia patients. Vitamin D levels were significantly lower in the β-thalassemia groups compared to controls (p = 0.0001) with 71.7% (n = 43/60) deficient despite 43.3% (n = 26/60) receiving supplements. Non-splenectomized patients had higher Procollagen Type I C-Peptide, a bone formation marker, compared to controls.
Conclusion:
Endocrine disturbances are common in β-thalassemia major despite chelation therapy. Incorporating endocrine assessment into routine practice is essential for early detection and management.
Plain language summary
β-Thalassemia major is a serious inherited blood disorder. Patients with this thalassemia type need regular blood transfusions to survive. Frequent blood transfusions lead to iron overload, which may damage organs including those involved in hormone production (the endocrine glands). Therefore, body growth, thyroid function, blood sugar level, bone strength, and overall well-being may be affected. This study involved 60 people with β-thalassemia major (7–35 years old) and 20 healthy people of similar age and gender. The goal was to see how hormone levels are affected in patients who are receiving iron removal treatments (chelation therapy) and to check if these problems are linked to iron levels in the body. Most patients (73%) had their spleen removed (splenectomy). Among them, 36 used deferiprone, 19 used deferasirox, and 5 used deferoxamine as iron chelators. Blood tests showed that both splenectomized and non-splenectomized patients had much higher iron and ferritin levels and lower haptoglobin compared to healthy people. Hepcidin and hemopexin levels were similar between groups. Concerning thyroid function, about 15% of patients had early signs of an underactive thyroid (subclinical hypothyroidism). Higher ferritin levels were linked to lower levels of free thyroxine, suggesting that excess iron may harm the thyroid. For calcium, parathyroid, and vitamin D levels, both underactive and overactive parathyroid glands were seen in about 11.7% of patients each. Vitamin D levels were much lower in the thalassemia group, and over 71.7% were deficient even though almost half were taking supplements. Bone health markers showed that non-splenectomized patients had higher levels of a bone formation protein (Procollagen Type I C-Peptide) compared to healthy controls. Regarding blood sugar control, the results were concerning. More than half of the patients had an HbA1c (a measure of long-term blood sugar) at or above the diabetes threshold, and almost 40% had impaired fasting blood sugar. Splenectomized patients had higher levels of fibroblast growth factor 21 (FGF21), a protein linked to metabolism and energy balance, compared to controls. Higher ferritin was associated with higher FGF21 levels. No major differences were found in galectin-1 or sortilin. In summary, hormone and metabolic problems were very common in people with β-thalassemia major, even though they were receiving treatment to reduce iron. These problems can affect many aspects of health, so regular hormone checks should be part of routine care to catch and treat issues early.
Keywords
Introduction
β-Thalassemia major is a disorder with a genetic mutation that leads to impaired production of hemoglobin β-globin chains.1,2 Thalassemia treatment involves regular blood transfusions and iron-chelation therapy to manage iron overload resulting from hemolytic anemia. Additional treatment options include hydroxyurea to increase the production of hemoglobin F, hematopoietic stem cell transplantation, and luspatercept, which enhances red blood cell production, thereby reducing the need for frequent transfusions and improving overall anemia management in patients with β-thalassemia. 3
At least one in every three to four children under the age of 12 with β-thalassemia major is affected by an endocrine disorder. The occurrence of these disorders is often associated with elevated ferritin levels and poor adherence to therapy. 4 Thalassemia-related endocrine disorders encompass hypogonadism, disturbances in the growth hormone and insulin-like growth factor 1 axis leading to growth retardation, hypothyroidism, hypoparathyroidism, abnormalities in glucose metabolism, adrenal insufficiency, decreased bone mineral density, and compromised bone microarchitecture, which increases the risk of fractures. 4 The prevalence of individual endocrine disorders varies depending on the population studied and the diagnostic criteria used. 4
The risk factors associated with endocrinopathies include iron overload—indicated by elevated serum ferritin levels—inadequate iron-chelation, history of splenectomy, and the early initiation of regular blood transfusions. 5 A study conducted in Turkey involving 31 transfusion-dependent β-thalassemia patients (9–23 years old) found that endocrine involvement was present in 83% of patients at the start of deferasirox iron-chelation therapy. This prevalence significantly decreased to 25.8% after an average follow-up period of 5.9 ± 2.02 years of treatment. 6 In a larger Italian multicenter longitudinal study conducted on 426 transfusion-dependent thalassemia patients receiving long-term deferasirox (median follow-up of 8 years), 121 out of 425 participants initially had one endocrine disorder, while 187 out of 426 had two or more. During follow-up, 104 new endocrine disorders were documented. Overall, it has been suggested that deferasirox therapy reduces the risk of endocrine disorders. 7
Concerning diabetes development, regular monitoring is advised for patients with β-thalassemia major to enable the early detection and effective management of associated endocrine complications. 8 For example, impaired glucose tolerance and insulin resistance typically develop before the onset of insulin deficiency and insulin-dependent diabetes mellitus. As a result, children with thalassemia receiving long-term transfusion and chelation therapy should undergo regular screening for glycemic disturbances starting after the age of 10. 9
There is a scarcity of studies evaluating the impact of deferiprone iron-chelation therapy on endocrine outcomes in β-thalassemia major. A 5-year adult cohort study, conducted in Italy, comparing different iron chelators (deferoxamine, deferiprone, and deferasirox), reported that deferiprone monotherapy, administered to 18 patients, did not lead to any significant improvement or reversal of endocrinopathies, including diabetes, hypothyroidism, and hypogonadism. 10 To address this gap, this case-controlled, cross-sectional study was conducted to assess endocrine complications in β-thalassemia major patients receiving deferiprone and deferasirox therapy. The study investigates the relationship between iron overload and hormonal or biochemical disturbances, with a focus on thyroid and parathyroid function, vitamin D status, metabolic markers, and bone turnover markers. In addition, comparisons are made between splenectomized patients, non-splenectomized patients, and healthy controls.
Materials and methods
Study design and participants
This case-controlled study included Jordanian patients of both sexes (7–35 years, 1:1 male to female ratio) admitted to the thalassemia and thrombophilia clinic at Zarqa Governmental Hospital and diagnosed with β-thalassemia major based on hemoglobin gel electrophoresis, with confirmation by high-performance liquid chromatography analysis. The study was approved by the Institutional Review Board (IRB) of the Ministry of Health, Amman, Jordan (IRB #138888). A participant’s written consent form was signed by the adults or the guardians. The control group consisted of sex- and age-matched healthy subjects (10–35 years). All cases had been diagnosed well in advance of the study. Participants were eligible only if they had maintained clinical stability during the 4 weeks prior to enrollment and had not undergone splenectomy within the past 6 months. Individuals were excluded if they had other forms of hemoglobinopathies, a prior history of bone marrow transplantation or gene therapy, significant impairment in renal function, ongoing infections (including hepatitis B, hepatitis C, or HIV), or demonstrated poor compliance with therapy or follow-up visits.
Clinical and laboratory parameters data were extracted from the electronic medical records including information about age, sex, date of diagnosis, splenectomy, medication, blood transfusion, calcium, glycosylated hemoglobin (HbA1c), fasting blood sugar (FBS), free thyroxine (FT4), thyroid-stimulating hormone (TSH), parathyroid hormone (PTH), vitamin D, ferritin, and iron levels. Patients with missing data were excluded from the study. The body mass index (BMI) was calculated by dividing body weight by the square of body height.
Blood sample collection
Whole peripheral blood samples (10 ml) were collected in plain tubes from participants. Then, serum concentrations for the following proteins were measured by using ELISA kits according to the manufacturer’s instructions. The following kits were used: Procollagen Type I C-Peptide (PICP; MBS2502579 Procollagen I C-terminal Propeptide; My BioSource, USA), Galectin-1 (Human Galectin-1 Kit catalogue No. EH203RB; Invitrogen ThermoFisher Scientific, USA), Sortilin (a neurotensin receptor family protein; Human Sortilin kit catalogue No. EH433RB; Invitrogen ThermoFisher Scientific, USA), Hepcidin (MBS2700551; My BioSource, USA), Haptoglobin (MBS2708032; My BioSource, USA), Hemopexin (HPX; MBS2704012; My BioSource, USA), and fibroblast growth factor 21 (FGF21; Human FGF21 Kit catalogue No. EH188RB; Invitrogen ThermoFisher, USA).
Statistical analysis
Data analysis was performed using SPSS version 26 (SPSS Inc., Chicago, IL, USA) for Windows. Categorical variables were summarized as frequencies and percentages, and numerical variables were expressed as means ± standard deviations. The Kolmogorov–Smirnov test was used to assess the normality of variable distributions, revealing that HPX was the only variable following a normal distribution. Normally distributed variables were analyzed using the one-way analysis of variance 11 followed by Tukey’s post hoc, whereas the Kruskal–Wallis test was applied to compare nonparametric variables, followed by the Bonferroni test. Two-tailed Spearman’s correlation was conducted, and predictors with significant correlations were further analyzed using simple linear regression.
Results
Demographic and clinical characteristics
Sixty β-thalassemia major patients (50% of them were males) and 20 sex- and age-matched healthy controls participated in the study. The ages of β-thalassemia patients ranged from 7 to 35 years, with 31 children (51.6%) under the age of 18 years. Splenectomized patients constituted 73.3% (n = 44/60) of the β-thalassemia group.
All β-thalassemia major patients received regular monthly blood transfusions. Post-transfusion furosemide, a diuretic, was administered to 98.3% (59/60) of them to prevent fluid overload and reduce cardiac complications. Its use was guided by clinical judgment and careful monitoring. In addition, all patients underwent iron-chelation therapy to manage transfusion-related iron overload: 36 received oral deferiprone (75–100 mg/kg/day), 19 received oral deferasirox (20–40 mg/kg/day), and 5 were treated with deferoxamine injection (20–40 mg/kg/day), with doses tailored according to body weight and iron burden. Moreover, three patients were taking metformin (850 mg, twice daily).
Iron overload in β-thalassemia major patients receiving iron-chelation therapy
Iron status, iron-regulating hormone (hepcidin), hemoglobin scavenging protein (haptoglobin), and heme scavenging protein (HPX) levels are shown in Table 1. A significant difference in iron, ferritin, and haptoglobin levels was found among the three studied groups, while no difference was observed in hepcidin or HPX levels (Table 1).
Iron status parameters.

Kruskal–Wallis test was applied to compare nonparametric variables followed by Bonferroni test.
P-values shown in bold represent statistically significant findings (p < 0.05).
HPX, hemopexin; SD, standard deviation.
Post hoc analysis showed that iron and ferritin levels were significantly higher in both splenectomized and non-splenectomized patients compared to the control group (p < 0.0001 for all), while no significant difference was found between the two thalassemia groups. For haptoglobin, both patient groups had significantly lower levels than controls, with p-values of <0.001 for splenectomized and <0.0001 for non-splenectomized individuals. In addition, a significant difference was observed between splenectomized and non-splenectomized patients (p = 0.012; Figure S1).
No significant differences were found for any tested parameter between patients taking different types of iron-chelation therapy, except for HPX. ANOVA showed that HPX levels were significantly lower in patients taking deferiprone (252.2 ± 71.6 µg/mL) compared to those on deferasirox (342.9 ± 68.9 µg/mL; p = 0.013). On the other hand, no significant difference was found in HPX levels between patients taking deferasirox and deferoxamine, or between those taking deferiprone and deferoxamine.
Effect of iron overload on thyroid function
In β-thalassemia major patients, none had overt hypothyroidism. However, 9 out of 60 of the β-thalassemia major patients (15%) had subclinical primary hypothyroidism compared to 0% in the control group. No significant differences in TSH, FT4 levels were found between splenectomized, non-splenectomized, and the control groups (Table 2).
Hormone and biochemical laboratory tests.

Kruskal–Wallis test was applied to compare nonparametric variables followed by Bonferroni test.
P-values shown in bold represent statistically significant findings (p < 0.05).
FBS, fasting blood sugar; FGF21, fibroblast growth factor 21; FT4, free thyroxine; HbA1c, glycosylated hemoglobin; PICP, procollagen type I C-peptide; PTH, parathyroid hormone; SD, standard deviation; TSH, thyroid-stimulating hormone.
In our study population, age, gender, and splenectomy were not correlated with FT4 or TSH levels. On the other hand, haptoglobin was weakly but significantly negatively correlated with TSH level (r = −0.263, p = 0.042), while ferritin was negatively and significantly correlated with FT4 (r = −0.330, p = 0.010; Figure 1). No significant correlation existed between iron, ferritin, hepcidin, or HPX with TSH. Similarly, no significant correlation existed between iron, haptoglobin, hepcidin, or HPX with FT4.
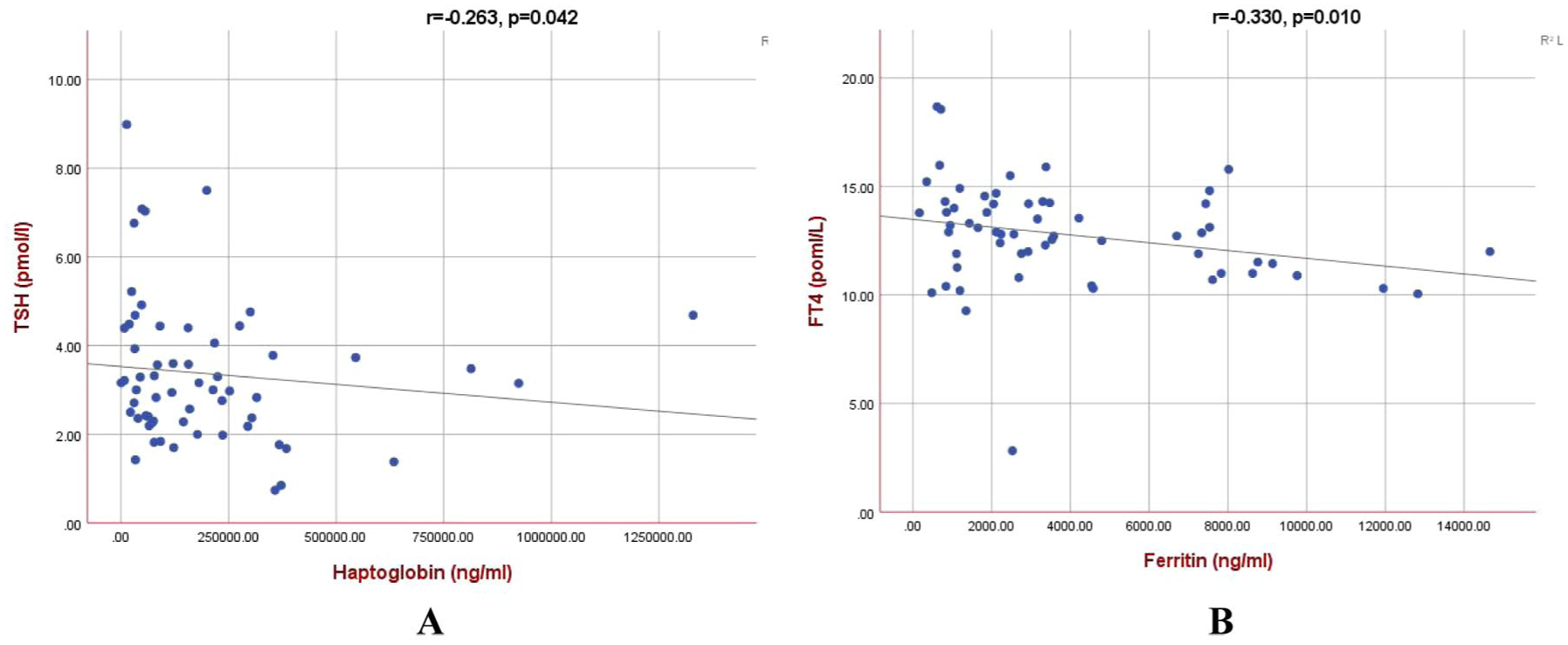
(a) Correlation between haptoglobin and TSH. (b) Correlation between ferritin and FT4.
Simple linear regression analysis revealed a statistically significant negative association between serum ferritin and FT4 levels. The unstandardized coefficient for ferritin was −0.0003 (standard error ≈ 0.0001), with a standardized β of −0.260 (t = −2.054, p = 0.044; 95% CI: −0.00048 to −0.00001). Although statistically significant, the effect size was minimal and may not be clinically meaningful. The model intercept was 13.484 (p < 0.001; 95% CI: 12.577–14.391). On the other hand, haptoglobin did not show a significant association with TSH levels (p = 0.367).
FBS, HbA1c, and metabolic regulation
According to our study, 51.7% (n = 31/60) of patients with thalassemia major had HbA1c ⩾6.5%, 15 out of 60 patients (25%) had HbA1c between 5.7% and 6.4%, and 38.3% (n = 23/60) of them had impaired fasting glucose. A significant difference between the three studied groups was found in FBS, HbA1c, and FGF21 but not in galectin-1 or sortilin levels (Table 2).
Post hoc analysis revealed a significant difference between the control group and the splenectomized group in FBS (p = 0.0001) as well as between the control and non-splenectomized group (p = 0.02). HbA1c levels were also significantly elevated in both splenectomized and non-splenectomized patients compared to the control group (p < 0.0001 for both). Also, a significant difference between the control group and the splenectomized group in FGF21 was found (p = 0.042), while the difference between non-splenectomized patients and controls did not reach statistical significance (Figure S2). The BMI was 21.23 ± 1.28, 21.93 ± 2.30, and 20.43 ± 3.24 kg/m2 for control, splenectomized, and non-splenectomized β-thalassemia groups, respectively, with no significant difference in BMI among the three groups.
FBS and FGF21 correlated significantly and positively with ferritin levels (r = 0.296, p = 0.022; r = 0.353, p = 0.006; respectively; Figure 2). Simple linear regression analysis showed that serum ferritin was a significant predictor of both FBS (p = 0.047) and FGF21 (p = 0.016). For FBS, the unstandardized coefficient (B) for ferritin was 0.006 (standard error = 0.003), indicating that for every 1 ng/mL increase in ferritin, the FBS level increased by approximately 0.006 mg/dL. This association was statistically significant (p = 0.047; 95% CI: 0.000–0.012). The standardized coefficient (β = 0.258) indicated a modest positive relationship between ferritin and FBS. The model intercept was 105.289 (p < 0.001; 95% CI: 75.527–135.051).
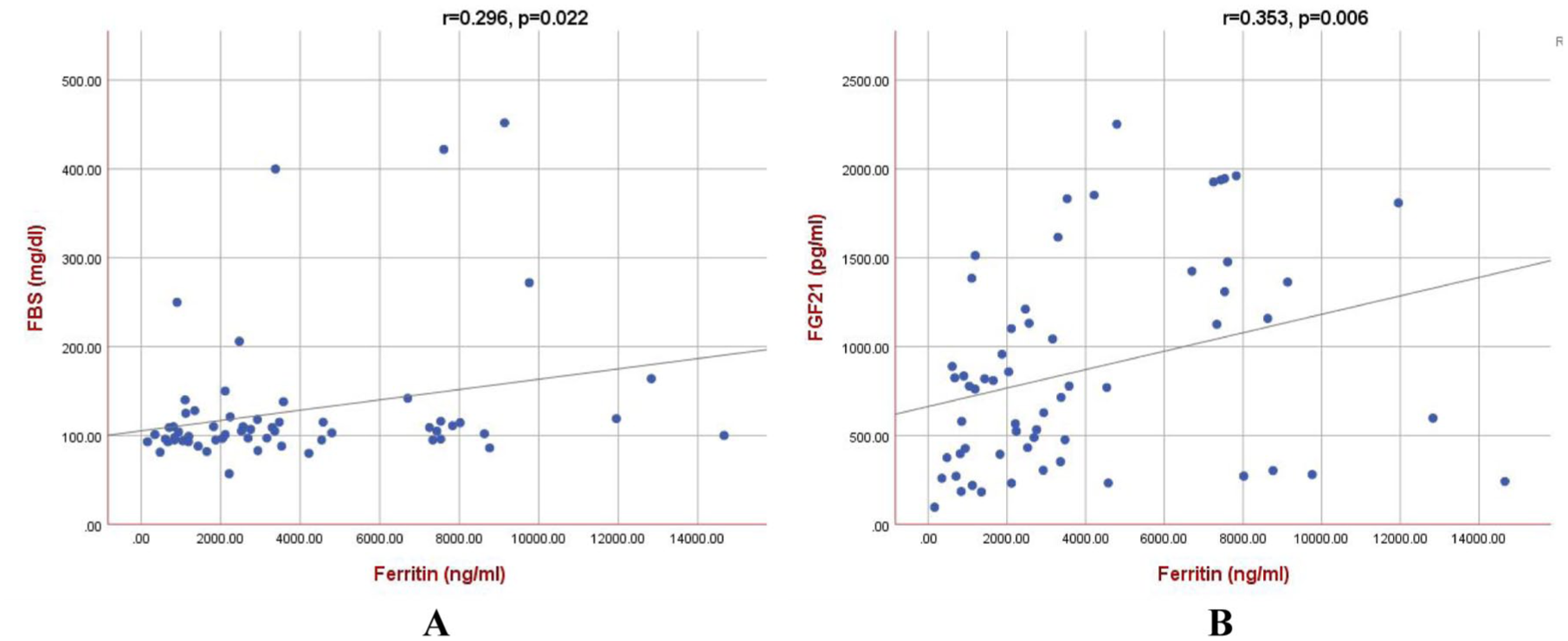
(a) Correlation between ferritin and FBS. (b) Correlation between ferritin and FGF21.
Similarly, a positive association was observed between ferritin and FGF21 levels. The unstandardized coefficient (B) for ferritin was 0.052 (standard error = 0.021), indicating that each 1 ng/mL increase in ferritin was associated with an approximate 0.052 pg/mL increase in FGF21. This relationship was statistically significant (p = 0.016; 95% CI: 0.010–0.094). The standardized coefficient (β = 0.309) suggested a moderate positive association between ferritin and FGF21. The model intercept was 663.586 (p < 0.001; 95% CI: 445.469–881.703).
PTH, vitamin D, and bone health
In this study of 60 patients with thalassemia major, 7 (11.7%) had hypoparathyroidism, 7 (11.7%) had hyperparathyroidism, and 43 (71.7%) had vitamin D deficiency, despite that 43.3% (26/60) of them received vitamin D supplementation. No significant difference in PTH levels was found between splenectomized, non-splenectomized, and the control groups, while a significant difference was found in vitamin D, calcium, and PICP, a biomarker of type I collagen synthesis (Table 2).
Post hoc analysis revealed that vitamin D levels were significantly lower in splenectomized and non-splenectomized patients compared to controls (p = 0.0001 for both), whereas only non-splenectomized patients had significantly elevated PICP compared to the control group (p = 0.014; Figure S3). In contrast, no statistically significant differences in calcium levels were observed between the groups after adjusting for multiple comparisons. While the unadjusted p-values initially indicated possible differences related to the splenectomy group, these findings did not remain significant following the Bonferroni correction.
Correlation analysis between ferritin level and PTH revealed no significant difference (r = 0.037, p = 0.299). Similarly, no significant correlation was found between ferritin level and vitamin D levels (r = −0.253, p = 0.051) or ferritin level and PICP (r = −0.014, p = 0.918; Figure 3).

(a) Correlation between PTH with ferritin. (b) Correlation between vitamin D with ferritin. (c) Correlation between PICP with ferritin.
Discussion
Iron status markers
Iron overload has long been recognized as a primary contributor to endocrine abnormalities in β-thalassemia 8 (Supplemental material). Serum ferritin, a reliable indicator of total body iron stores, is commonly used to assess iron status. 12 This study evaluated some endocrine abnormalities in β-thalassemia major patients on iron-chelation therapy and studied their association with ferritin.
β-Thalassemia major patients had marked iron metabolism disturbances, with significantly elevated serum iron and ferritin levels compared to controls, while no significant differences were found between splenectomized and non-splenectomized patients. These observations are consistent with a prior report from Egypt indicating that splenectomy does not lessen the iron burden in children with thalassemia since the ferritin levels remained high after the procedure. 13
Despite iron overload, hepcidin levels were not significantly different among the groups. This may seem paradoxical, as hepcidin usually increases with iron excess. It is well known that ineffective erythropoiesis in β-thalassemia suppresses hepcidin despite high ferritin, promoting continued iron absorption and overload. This is mainly due to the elevated erythroferrone and growth differentiation factor 15 levels, which inhibit hepcidin by disrupting BMP/SMAD signaling.14,15 In addition, other erythroid factors and cytokines such as IL-6 may also contribute to this dysregulation. 16
Haptoglobin levels were markedly lower in both thalassemia groups compared to controls. This reflects ongoing hemolysis and saturation of its binding capacity, which is a hallmark of β-thalassemia. These findings align with a previous study from Egypt, which reported that systemic iron overload in β-thalassemia major is associated with decreased haptoglobin levels in patients 12–20 years old. 17 On the other hand, hemopexin levels were not significantly different from the controls in contrast to a previous study performed on a limited number of children over 3 years (n = 3) in the USA that reported reduced hemopexin levels in β-thalassemia major. 18 One possible explanation for these findings is that in chronic hemolytic states such as β-thalassemia, continuous low-grade hemolysis may result in ongoing hemopexin consumption and replenishment, keeping its levels within a relatively stable range 19 while the reduction in haptoglobin levels is attributed to its rapid binding of free hemoglobin released during hemolysis, followed by swift clearance from circulation, leading to its depletion. In contrast, hemopexin binds free heme more slowly and is continuously replenished by the liver, allowing its levels to remain relatively stable despite ongoing hemolysis. It is also important to note that, unlike haptoglobin, hemopexin is not an acute-phase protein in humans, which further contributes to the stability of its serum concentration. 19 Future studies stratifying patients by transfusion status, variability in hemolysis rate, and inflammatory parameters may provide greater clarity on these findings.
Most patients in this study were receiving deferiprone therapy. This drug was traditionally considered a second-line chelator, but it is often used as a first-line treatment in our clinical setting due to its oral administration, better patient compliance, and proven efficacy in reducing cardiac iron. In addition, some patients may have been intolerant to or inadequately managed with deferoxamine, which requires parenteral administration and is often associated with poor adherence. Therefore, the choice of an iron-chelation drug reflects both patient-specific factors and institutional treatment protocols. While deferasirox offers the convenience of once-daily oral dosing and is typically associated with better compliance than deferiprone, its higher cost and limited availability in some healthcare settings may restrict its widespread use. In addition, local treatment guidelines, physician preference, and differences in side effect profiles between chelators can further influence therapy selection. Overall, the predominance of deferiprone use in this cohort likely reflects a multifactorial decision-making process shaped by clinical efficacy, patient tolerance, economic considerations, and institutional practices.
No significant differences were found in any tested parameter between patients taking different types of iron-chelation therapy, except for HPX, which was significantly lower in patients taking deferiprone than those on deferasirox. This finding is challenging to interpret without accounting for potential confounding factors such as drug dose and other variables. Further studies are necessary to determine whether the type of iron-chelation therapy influences HPX levels.
Thyroid dysfunction
In the present study, approximately 15% (9/60) of β-thalassemia major patients receiving iron-chelation therapy had subclinical hypothyroidism, characterized by elevated TSH levels with normal FT4. Notably, none of the participants had overt hypothyroidism. These findings are consistent with an earlier study conducted in the United Arab Emirates, reporting elevated TSH in 14.7% of children and 8.1% of adolescents, whereas low FT4 levels were detected in only one child and one adolescent among the patients with β-thalassemia major. 20 On the other hand, several studies have reported a lower prevalence of subclinical hypothyroidism among patients with β-thalassemia major, including 7.22% (7 out of 97) in Pakistani children, 21 12.8% (9 out of 70) in a mixed cohort of Turkish children and adults, 8 and 5% (11 out of 120) in Egyptian children. 22 Surprisingly, a recent study conducted in Thailand reported an exceptionally high prevalence of hypothyroidism (47.8%) in patients with hemoglobin E/β-thalassemia, including both children and adults, with subclinical hypothyroidism observed in 63.6% of the cases. 5
In the present study, ferritin was a significant predictor of FT4. These findings are consistent with other studies reporting that elevated serum ferritin levels and poor treatment compliance were significantly associated with a higher risk of endocrine complications.8,22 It has been reported that endocrinopathies are frequently observed in thalassemia major patients despite deferoxamine therapy, particularly among those with serum ferritin levels exceeding 2500 μg/L or those who have undergone splenectomy. 23
Gender showed no significant correlation with FT4 or TSH levels in the present investigation, consistent with the findings of a recent study conducted in Pakistan and involving only children with β-thalassemia. 21 Concerning splenectomy, our findings are consistent with a study from Greece, which suggested that splenectomy may not significantly influence the development of hypothyroidism in transfusion-dependent thalassemia patients (both children and adults). 24 However, they contrast with a study from the United Arab Emirates (including children and adults), which reported a higher incidence of hypothyroidism in splenectomized patients (26%) compared to non-splenectomized patients (4.5%). 23
The prevalence of hypothyroidism in β-thalassemia major varies among studies due to several factors. Age and duration of transfusion therapy play key roles since older patients with prolonged transfusions have higher iron overload, increasing thyroid risk. 20 Other factors include variations in therapeutic protocols and iron-chelation therapy, 10 the mean age at which transfusion was started, 25 splenectomy, 23 bone marrow transplantation, 5 and differences in thyroid function testing methods and diagnostic thresholds.
The findings of different studies underscore the critical importance of routine thyroid function screening for early detection, as well as the need for long-term, regular monitoring in thalassemic patients. The management of hypothyroidism in thalassemia should be individualized. Asymptomatic patients with TSH < 10 mIU/L may be monitored, while L-thyroxine is advised for symptomatic cases or TSH ⩾ 10 mIU/L due to risks to growth and development. 26 Subclinical hypothyroidism may progress, especially with iron overload or inadequate chelation; therefore, close follow-up is essential.
Glucose dysregulation
Diabetes in β-thalassemia major patients is different from both type 1 and type 2 diabetes, though it shares some features with each. It is usually diagnosed using high FBS, high random blood sugar with symptoms, or a diabetic response to an oral glucose tolerance test. 27 In the current study, both splenectomized and non-splenectomized patients showed significantly higher HbA1c levels compared to the control group, with no significant difference between the two patient groups. In addition, approximately 51.6% of β-thalassemia major patients had HbA1c levels above 6.5%, aligning with the findings from China that HbA1c increases with the severity of glucose metabolism abnormalities in β-thalassemia major children. 28 Traditional HbA1c thresholds may be unreliable in β-thalassemia due to hemolysis and abnormal hemoglobin, which shortens red blood cell lifespan. Frequent transfusions also affect HbA1c accuracy, which can lead to falsely elevated or declined HbA1c levels. Overall, standard diagnostic thresholds must be interpreted with caution in these patients. 29 Although the reliability of HbA1c in β-thalassemia major is still debated, it continues to serve as a valuable marker for identifying glucose dysregulation. 29 The high prevalence of insulin resistance among patients with β-thalassemia emphasizes the importance of regular monitoring of their glycemic status.
In this study, 38.3% of β-thalassemia major patients (including children and adults) had impaired fasting glucose. Both splenectomized and non-splenectomized patients had higher FBS levels than healthy controls, but there was no significant difference between them. Studies have reported widely varying rates of glucose abnormalities in patients with β-thalassemia major. A study from Egypt found a lower prevalence of impaired glucose tolerance identified in 4.17% and impaired fasting glucose in 3.33%, with no cases of diabetes in children with β-thalassemia. 22 Another study from Turkey reported moderate rates, with diabetes in about 10% of patients, including children and adults, and impaired glucose tolerance in 7.1%. 8 In contrast, higher rates have also been observed, with diabetes affecting up to 41% of adults with β-thalassemia from the UK. 30 In a meta-analysis that included 44 studies comprising a total of 16,605 cases of β-thalassemia major, the overall prevalence of diabetes mellitus was 6.54%. Subgroup analysis based on geographic region showed the highest prevalence in the Middle East, at 7.90%. 31 Endocrine complications were more frequent in diabetic patients, with higher rates of hypothyroidism (19% vs 10%) and hypoparathyroidism (21% vs 5%) compared to nondiabetics. 32 The differences in the prevalence of diabetes among studies may be due to variations in age, iron overload, disease severity, or diagnostic methods across studies.
Iron overload and liver dysfunction have been identified as key contributors to glucose metabolism disturbances in patients with β-thalassemia major. 27 Both baseline and follow-up levels of serum ferritin and alanine aminotransferase were found to be independent predictors of progression to diabetes mellitus. 33 In the current study, very high ferritin levels were present in β-thalassemia patients despite iron-chelation therapy. This may explain the relatively high percentage of patients with glucose dysregulation. In clinical practice, it is difficult to give a definitive cut-off level of serum ferritin to define patients at risk of glucose dysregulation. 34 Interestingly, efficient iron-chelation monotherapy in patients with β-thalassemia major and keeping serum ferritin levels less than 1000 ng/mL did not entirely prevent glucose metabolism disorders, including abnormalities of insulin secretion and sensitivity. 33
FGF21, galectin-1, and sortilin were studied due to their involvement in metabolic regulation. Galectin-1 and sortilin levels did not differ significantly between the control and β-thalassemia groups. In contrast, FGF21 levels were significantly higher in the splenectomized group compared to controls. In addition, FGF21 showed a significant positive correlation with ferritin, and linear regression analysis identified ferritin as a significant predictor of FGF21 levels. While correlation and regression quantify associations, they do not establish causation without controlling for confounders or using experimental designs. However, the association between FGF21 and ferritin is noteworthy and may reflect subclinical organ stress in β-thalassemia, where iron overload leads to oxidative damage and mitochondrial dysfunction. 35 As a liver-derived stress hormone, elevated FGF21 could serve as a biomarker of iron-induced tissue injury, complementing traditional markers like ferritin. Monitoring its levels may aid in risk assessment and guide therapeutic decisions.
Vitamin D, parathyroid function, and bone health
Among patients with β-thalassemia major, 71.67% (43/60) had vitamin D deficiency, despite that 43.3% (26/60) were receiving vitamin D supplementation. At the same time, 11.67% (7/60) of patients had hyperparathyroidism, which is considerably higher than what was reported earlier from Turkey (2.8% including children and adults), Egypt (6.66% including only children), but lower than what was reported in another study from Turkey (25.6% including only children).8,22,36 Serum ferritin was not correlated with PTH levels in our study, which aligns with the findings of a study from Indonesia, involving both children and adults. 37
The elevated PTH levels observed in our study are most likely a compensatory response to vitamin D deficiency. It is well established that vitamin D supports bone health by enhancing calcium and phosphate absorption and promoting proper bone mineralization. In the context of vitamin D deficiency, intestinal calcium absorption is impaired, leading to a tendency toward hypocalcemia. To maintain normocalcemia, the parathyroid glands increase the secretion of PTH, resulting in secondary hyperparathyroidism characterized by elevated PTH levels. Sustained elevation of PTH accelerates bone resorption and contributes to bone loss, as reported earlier, where a negative correlation between bone mineral density and PTH levels exists. Abnormal bone mineral density was observed in 42.9% of women and 23.1% of men with β-thalassemia major. 38
The persistently high prevalence of vitamin D deficiency in this cohort, despite supplementation, is likely multifactorial. Malabsorption may contribute, particularly in thalassemia patients with iron-induced gastrointestinal or pancreatic dysfunction, which can impair the absorption of fat-soluble vitamins. Hepatic dysfunction is a common consequence of iron overload and may reduce the conversion of vitamin D to its active form via impaired 25-hydroxylation. Inadequate dosing relative to the patients’ metabolic demands and altered pharmacokinetics may also play a role. In addition, the use of furosemide (a diuretic) and calcium carbonate supplements can increase urinary calcium excretion and disrupt calcium-vitamin D homeostasis.39,40 Reduced sunlight exposure and genetic polymorphisms influencing vitamin D metabolism also contribute further to the condition. Moreover, vitamin D deficiency may reflect a broader regional problem. A recent study of vitamin D status in Jordanian adults reported an alarmingly high prevalence of low vitamin D levels, with 89.7% having serum vitamin D concentrations below 30 ng/mL, underscoring a widespread public health concern. 41
PICP is a sensitive biomarker of bone formation. It is released into the bloodstream during the synthesis of type I collagen, which is the main collagen found in bone. In this study, the non-splenectomized thalassemia patients had significantly higher PICP levels compared to the control. In thalassemic patients, especially those with β-thalassemia in early stages or on regular transfusions, elevated or normal PICP levels are seen initially due to high bone turnover. This reflects active but often ineffective bone formation occurring alongside heightened bone resorption, which is a characteristic feature of thalassemic osteopathy. As bone formation capacity declines over time, PICP levels tend to decrease. Persistent high turnover disrupts bone mineralization and, therefore, raises the risk of osteoporosis. Therefore, monitoring PICP along with bone resorption markers offers a comprehensive assessment of bone remodeling and aids early detection to prevent long-term skeletal complications. 42
This study has several limitations that should be acknowledged. First, the relatively small sample size may limit the statistical power to detect significant associations. Second, the cross-sectional design restricts the ability to establish causal relationships between iron overload and endocrine dysfunction. While correlations can be observed, the temporal sequence of events and whether iron overload leads to endocrine disturbances or vice versa cannot be determined without longitudinal follow-up. Therefore, larger multicenter, longitudinal studies are recommended.
Conclusion
This case-control study provides new insights into the endocrine and metabolic disturbances in β-thalassemia major patients. It underscores the high prevalence of subclinical hypothyroidism, impaired glucose metabolism, altered bone remodeling, and marked vitamin D deficiency despite iron-chelation therapy. These findings emphasize the importance of improving monitoring and personalized management, including optimization of iron chelation, to prevent complications and improve patient outcomes.
The results of the study revealed many research gaps in β-thalassemia management. It stresses the need to standardize vitamin D supplementation, considering drug interactions and kidney function. It also shows that HbA1c is unreliable for diabetes monitoring in these patients, urging the need for better markers. In addition, it highlights FGF21 as a potential biomarker for iron-induced tissue damage, especially in the liver and endocrine glands, and this aspect requires further investigation.
Supplemental Material
sj-doc-3-tae-10.1177_20420188251406554 – Supplemental material for Endocrine complications in patients with β-thalassemia major receiving iron-chelation therapy
Supplemental material, sj-doc-3-tae-10.1177_20420188251406554 for Endocrine complications in patients with β-thalassemia major receiving iron-chelation therapy by Ola M. Al-Sanabra, Manal A. Abbas, Abeer A. Hazàa and Wafà J. Hazà in Therapeutic Advances in Endocrinology and Metabolism
Supplemental Material
sj-docx-1-tae-10.1177_20420188251406554 – Supplemental material for Endocrine complications in patients with β-thalassemia major receiving iron-chelation therapy
Supplemental material, sj-docx-1-tae-10.1177_20420188251406554 for Endocrine complications in patients with β-thalassemia major receiving iron-chelation therapy by Ola M. Al-Sanabra, Manal A. Abbas, Abeer A. Hazàa and Wafà J. Hazà in Therapeutic Advances in Endocrinology and Metabolism
Supplemental Material
sj-docx-2-tae-10.1177_20420188251406554 – Supplemental material for Endocrine complications in patients with β-thalassemia major receiving iron-chelation therapy
Supplemental material, sj-docx-2-tae-10.1177_20420188251406554 for Endocrine complications in patients with β-thalassemia major receiving iron-chelation therapy by Ola M. Al-Sanabra, Manal A. Abbas, Abeer A. Hazàa and Wafà J. Hazà in Therapeutic Advances in Endocrinology and Metabolism
Footnotes
Acknowledgements
The authors gratefully acknowledge Dr Ahmad Khairi/Zarqa Governmental Hospital for his valuable assistance in providing detailed information regarding the treatment protocols for β-thalassemia patients.
Declarations
Ethics approval and consent to participate
The study was approved by the Institutional Review Board (IRB) of the Ministry of Health, Amman, Jordan (IRB #138888). A participant’s written consent form was signed by adults or children’s guardians.
Consent for publication
Not applicable.
Author contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Competing interests
The authors declare that there is no conflict of interest.
Availability of data and materials
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.