
Abstract
Cardiovascular disease is the leading cause of death worldwide; however, women tend to be less affected than men during their reproductive years. The female cardiovascular risk increases significantly around the time of the menopausal transition. The loss of the protective action of ovarian oestrogens and the circulating androgens has been implicated in possibly inducing subclinical and overt changes in the cardiovascular system after the menopausal transition. In vitro studies performed in human or animal cell lines demonstrate an adverse effect of testosterone on endothelial cell function and nitric oxide bioavailability. Cohort studies evaluating associations between testosterone and/or dehydroepiandrosterone and subclinical vascular disease and clinical cardiovascular events show an increased risk for women with more pronounced androgenicity. However, a mediating effect of insulin resistance is possible. Data on cardiovascular implications following low-dose testosterone treatment in middle-aged women or high-dose testosterone supplementation for gender affirmatory purposes remain primarily inconsistent. It is prudent to consider the possible adverse association between testosterone and endothelial function during the decision-making process of the most appropriate treatment for a postmenopausal woman.
Introduction
Cardiovascular disease (CVD) remains the leading cause of death worldwide, accounting for 30.3% of women’s all-cause mortality in the United States in 2018.1,2 Although incidence and mortality of CVD, in general, are lower in women compared to men, estimated as 25% lower risk for incident CVD and 38% lower risk for all-cause mortality, this trend tends to reverse after midlife. 3 Data from the NHANES (National Health and Nutrition Examination Survey) from 2015 to 2018 show that CVD prevalence is estimated at 75.4% for women aged 60–79 years and up to 90.8% for women aged more than 80 years, incorporating hypertension as a primary cardiovascular outcome. 2 Interestingly, coronary heart disease in women notably increases during midlife,2,4 coinciding with the menopausal transition. 5
The age of ovarian senescence appears as an additional important determinant of overall cardiovascular risk. In a pooled analysis of 203,767 postmenopausal women, 6 retrieved from observational studies, an inverse-graded association was observed between younger age at natural or surgical menopause and rates of incident CVD. The risk has been estimated as 1.59-times higher in women experiencing menopause at an age less than 35 years as opposed to women experiencing menopause at the age of 50–54 years. 6 In addition, women experiencing early menopause or premature ovarian insufficiency had a higher relative risk for prevalent coronary heart disease and CVD mortality, estimated as 1.5-times and 1.19-times higher, respectively, compared with women experiencing spontaneous menopause at an age more than 45 years. 7 Similarly, women experiencing menopause at the age of 50–54 years had a 13% lower risk of fatal coronary heart disease than women experiencing menopause at 50 years. 7
According to the latest statement of the American Heart Association, the menopausal transition is an essential window of accelerated cardiovascular risk suggesting the acquisition of primary prevention strategies. 5 Recent evidence highlighted the importance of both the traditional risk factors and additional sex-specific factors, including the possible effect of sex hormones in determining the overall cardiovascular risk. 8 A large amount of data support a possible association between hormonal changes associated with menopause and age-related alterations of the cardiovascular system, further contributing to the sharp increase of CVD observed at midlife.5,8 However, the available literature seems to be characterized by heterogeneity, determined by the study design.
The aim of this review is to summarize the evidence on the association between androgens and subclinical as well as clinical CVD in ageing women.
Cardiovascular ageing: the effect of menopause
The process of cardiovascular ageing is comprised of a series of changes affecting both the left ventricle as well as the overall arterial tree. The effect of ageing on the left ventricle seems to range from concentric hypertrophy with or without diastolic dysfunction, aortic valvulopathy, reduced variability of the heart rate, and annular calcification of the mitral valve as well as conduction abnormalities. 9 The arterial tree itself is characterized by progressively enlarging vascular lumen as well as thickening of the vessel wall.9,10 Simultaneously, early stages of vasodilation become apparent in the aorta, the large branches proximal to the myocardium.9,10 The raised aortic stiffness results in a further rise in reflected waves and aortic pulse wave velocity, which became apparent as elevated systolic blood pressure and increased left ventricular afterload. 10 Diastolic blood pressure, on the contrary, tends to decline with age. 10 The raised systolic blood pressure in the context of low diastolic blood pressure results in increased oxygen demand by the myocardium, the supply of which is likely to be compromised in the context of advancing atherosclerosis of coronary arteries. 10
At a cellular level, ageing endothelial cells and cardiomyocytes produce increasing amounts of reactive oxygen species (ROS).11–13 This process takes place principally in the mitochondria, which constitute up to 45% of the cardiac cell volume.14,15 Ultimately, ageing-mediated increased production of ROS compromises vasorelaxation, increases vascular stiffness and impairs endothelial cell turnover and release of nitric oxide.16,17 At a cellular level, oxidative stress and marked production of ROS result in the following changes, which contribute to the development of heart failure, valvular degeneration, and atrial fibrillation: (1) epigenetic alterations and DNA damage, (2) increased expression of tumour necrosis factor-alpha and matrix metalloproteinase’s, (3) impaired function of the sarcoplasmic/endoplasmic reticulum calcium-ATPase and consequently insufficient reuptake of ionized calcium, (4) impaired bioavailability of nitric oxide and suppressed function of endothelial nitric oxide synthase.16,17
Menopause-associated hormone changes contribute to the age-related alterations evident in the cardiovascular system. Oestrogen deficiency is implicated in endothelial dysfunction and reduced NO bioavailability. 18 The postmenopausal ovary continues to produce testosterone, the levels of which decline progressively with ageing.19,20 The adrenal glands contribute to the pool of testosterone through an ongoing secretion of dehydroepiandrosterone sulphate (DHEAS), the levels of which also decline with ageing, as well as androstenedione.19,20 Androgen production during the female reproductive life is evident as follows: 20 (1) in the ovarian theca cells, which are secreting up to 20% of circulating DHEAS, (2) in the ovarian stroma, which is producing 50% of the circulating androstenedione and up to 25% of the circulating testosterone, (3) the adrenal zona reticularis which is secreting up to 50% of circulating DHEAS, androstenedione and up to 25% of circulating testosterone. The remaining portion of testosterone is produced with the direct conversion from circulating androstenedione. 20
Increasing age, as well as the menopause-related hormonal changes, result in changes that directly or indirectly contribute to increase in cardiovascular risk 21 (Figure 1). Following ovarian senescence, the production of oestrogen decreases rapidly. 22 Simultaneously, levels of sex hormone-binding globulin also decrease due to the subsequent reduction in oestrogen-liver interaction, 23 a change that results in an increase in circulating levels of sex hormones. 24 The rate of androgen production declines to a lesser extent, given the ongoing contribution of the adrenal glands to the pool of circulating sex hormones. 20 Consequently, the androgen-to-oestrogen ratio increases, resulting in an environment of relative androgenicity. 25 These hormonal alterations, as well as ageing per se, are known to induce pathophysiological manifestations, which consist of impaired fibrinolysis, 26 insulin resistance, 27 and visceral adiposity. 28 In this context, impaired fibrinolysis contributes to the origin and progression of atherosclerotic changes.29,30 Hyperandrogenism, visceral adiposity, and insulin resistance contribute to metabolic dysfunction,31,32 generation of oxidative stress and low-grade inflammation,33,34 which have been linked with endothelial dysfunction. 35 As described by experimental data, the early stages of the atherosclerotic process, as well as hyperandrogenemia, have also been demonstrated to contribute to endothelial dysfunction,36,37 which eventually results in more advanced atherosclerotic changes. 38 Endothelial dysfunction is a recognized factor promoting the development of atherosclerosis. 39

Pathophysiology of menopausal hormone changes and the related effects on the vascular endothelium.
The effect of androgens on cardiovascular tissues: preclinical studies
Androgen receptors are widely distributed in different cell types of the cardiovascular tissue; their presence has been confirmed in endothelial cells, cardiomyocytes and vascular smooth muscle cells (VSMCs). 40 The interaction between androgens and endothelial cells is of particular significance, as these represent key players in cardiovascular pathophysiology, with a fundamental role in cardiovascular health and disease. 41
Emerging data from in vitro studies suggest cardiovascular implications of testosterone to cells of the cardiovascular system (Table 1). Overall, animal studies support that testosterone improves endothelial mediated vasodilation,42,47 increases ROS generation and oxidative stress,46,60 and promotes apoptosis and migration of VSMCs.48,49 Data on angiogenesis are conflicting, with evidence supporting a positive effect of testosterone in male animals only, with no effect in females. 45 The results on a possible link between testosterone and VSMC calcification remain conflicting, with studies supporting stimulation of VSMCs migration, 48 induction of VSMCs-apoptosis, 49 or suppression50,51 of VSMCs-calcification. Studies addressing the effect of testosterone on cardiomyocytes report protection from apoptosis, 52 but also induction of apoptosis, 54 suppression of oxidative stress 55 and increased intracellular calcium ion signalling. 53 Moreover, preclinical studies describe the effect of the synergistic action of estradiol and testosterone on cells of the cardiovascular system. Accordingly, spontaneously hypertensive ovariectomized rats treated with combined equine oestrogen (CEE) showed suppression of oxidative stress (ROS). 56 This beneficial effect of CEE-treatment was reversed after the addition of testosterone, with induction of ROS.56,57 Others reported that the optimal estradiol to testosterone ratio following treatment with a combination of estradiol and testosterone is associated with a decrease in apoptosis,58,59 endothelial injury, vascular inflammation, coagulation, and formation of foam cells and vascular lipid lesions. 59
Experimental data on the effect of testosterone on cells of the cardiovascular system.

A, androgens; AR, androgen receptor; cAMP, cyclic adenosine monophosphate; CEE, conjugated equine oestrogen; CP4 F3, cytochrome P450 4F3B; DHT, dihydrotestosterone; EC, endothelial cells; GSH-Px, glutathione peroxidase; HUVEC, human umbilical vein endothelial cells; iPSC-CMs, pluripotent stem cell-derived cardiomyocytes; L-VOCCs, L-type voltage operated Ca 2 + channels; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; mtDNA, mitochondrial deoxyribonucleid acid; NADPH, Nicotinamide Adenine Dinucleotide Phosphate; PAI-I, plasminogen activator inhibitor I; PI3 K/Akt, phosphoinositide-3-kinase/protein kinase B; PTX, pertussis toxin; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; NO, nitric oxide; PKC, protein kinase C; ROS, Reactive oxygen species; SOD, superoxide dismutase; Src, regulator of redox-sensitive migration; T, testosterone; t-PA, tissue plasminogen activator; VSMC, vascular smooth muscle cell; (↑), tendency to increase; (↓), tendency to decrease; 20-HETE, 20-hydroxyeicosatetraenoic acid.
Data from preclinical studies describing the effect of dehydroepiandrosterone (DHEAS) on cardiovascular tissues are presented in Table 2. Experimental data on the effect of DHEAS administered to endothelial cells (ECs) describe that DHEAS-treatment is reducing the proliferation rate, 61 or increasing the proliferation rate of ECs.67,69 DHEAS has, furthermore, been reported to induce endothelial cell necrosis.61,64 Others described reduction of vascular inflammation, 62 exertion of antioxidative and anti-inflammatory effects,65,66 increased angiogenesis, 67 and improved endothelial function. 68 DHEAS treatment administered to VSMCs results in reduced inflammation and oxidative stress in healthy VSMCs. 71 In studies describing balloon injury models, DHEAS treatment is linked with reduced VSMC migration and proliferation72,73 and increased apoptosis. 72 The effect of DHEAS-treatment on cultures of cardiomyocytes consists of a tendency towards a lower chronotropic and hypertrophic response, 76 reduced pulmonary artery pressure, prevention of pulmonary artery hyperreactivity and remodelling and prevention of the remodelling of the right ventricle. 74
Experimental data on the effect of DHEAS on cardiovascular tissues.

BKCa, Ca2+-activated potassium channel; BNP, brain natriuretic peptide; DHEA, dehydroepiandrosterone; DHP-CCBs, dihydropyridine Ca2+ channel blockers; EC, endothelial cells; ERK1/2, extracellular signal-regulated kinase 1/2; G6PD, glucose-6-phosphate dehydrogenase; HF, heart failure; HUVEC, human umbilical vein endothelial cells; ICAM, 1, intercellular adhesion molecule 1; LDL, low-density lipoprotein; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemoattractant protein-1; MDA, malondialdehyde; mRNA, messenger rivonucleid acid; NCAM, neural cell adhesion molecule to monocyte; NO, nitric oxide; NPs, nanoparticles; PI3 K, phosphatidylinositol 3-kinase; PM, particulate matter; PTX, pertussis toxin; ROS, reactive oxygen species; RV, right ventricle; TNF, tumour necrosis factor; VCAM, vascular adhesion molecule; (↑), tending to increase; (↓), tending to decrease; 6-AN, 6-aminonicotinamide.
Pathophysiological mechanisms determining the interaction between menopause and endothelial cells
Endothelial cells are known to express both oestrogen (ER) and androgen receptors (AR), which are known to mediate their functional capacity. 40 The binding of oestrogen on the ER has been linked with decreased expression of adhesion molecules (e.g. CD40, P-selectin, E-selectin, ICAM-1) as a response to atherogenic promoting factors, ensuing in increased NO release and VSMC relaxation. 78 The chronic oestrogen deficiency following ovarian senescence contributes to endothelial dysfunction with a decrease in NO bioavailability, secondary to decreased NO synthesis and increased NO inactivation. 79 Moreover, the postmenopausal environment of hypoestrogenemia induces an altered redox balance, contributing to oxidative stress, 80 while also inducing the expression of homocysteine and cysteine molecules, with adverse effects on endothelial function. 81
Focusing on the interactions mediated by androgens, testosterone has been linked with a dual effect on cardiovascular cells, both pro-oxidant and antioxidant. 60 More specifically, the pro-oxidant effect consists of suppressed activation of the endothelial NO synthase (eNOS), resulting in impaired endothelial NO release in women.37,82 The antioxidant effect is maintained through conversion to estradiol, which also contributes to mediating high levels of antioxidant enzymes. 60 Earlier data suggested that the degree of oxidative stress is an essential factor regulating testosterone action; thus, an environment of high oxidative stress is related to adverse testosterone effects and vice versa. 55 These observations highlight the need for further research to determine factors driving oxidative stress during and after the menopausal transition.
Data on the cardiovascular alterations observed in women during the menopausal transition is rather sparse. Samargandy et al. 83 evaluated 339 women retrieved from the Study of Women’s Health Across the Nation, aiming to measure the annual percentage change in carotid-femoral PWV in relation to the final menstrual period (FMP). This study 83 highlighted the pattern of statistically significant changes in values of cfPWV around the FMP. Accordingly, cfPWV values increased at a steady rate up to 12 months prior to the FMP (0.9%, 95% CI: −0.6% to 2.3%), continued to increase more rapidly up to 1 year after the FMP (7.5%, 95% CI: 4.1% to 11.1%), and subsequently decreased at a steady rate (–1.0%, 95% CI: −2.8% to 0.8%).
Endogenous androgens and CVD: data from human cohort studies
Subclinical CVD
Data on cohort studies assessing the possible associations between endogenous androgens and markers of subclinical vascular disease are presented in Table 3.
Cohort studies exploring the possible associations between endogenous androgens and subclinical vascular disease.

AAD, ascending aortic distensibility; CAC, coronary artery calcification; CARDIA, coronary artery risk development in young adults; CAS, cumulative marker combining pulse wave velocity and stiffness index; CI, confidence interval; CS, cross-sectional; CVD, cardiovascular disease; FAI, free androgen index; FT, free testosterone; FU, follow-up; IMT, intima media thickness; m, months; max, maximum; NA, non applicable; NRI, net reclassification index; NS, non-significant; OR, odds ratio; PWV, pulse wave velocity; SHBG, sex hormone binding globulin; SIindex, stiffness index; TT, total testosterone; y, years.
The possible association between endogenous androgens and endothelial function in middle-aged women has been investigated by one cohort study. Georgiopoulos et al. 84 evaluated endothelial function in a total of 180 postmenopausal women with no overt CVD or diabetes during a follow-up period of up to 29 months. This study concluded that the pattern of change in %FMD levels during the follow-up period was associated with baseline levels of FAI: one SD increase in levels of baseline FAI was linked with a 0.42% decrease in mean FMD values, measured during follow-up.
The possible association between endogenous androgens and markers of arterial stiffness in middle-aged women has been investigated by three cohort studies. Subramanya et al. 91 investigated 1345 postmenopausal women aged 45–84 years, retrieved from the MESA (multi-ethnic study in atherosclerosis), describing evidence of cross-sectional but not longitudinal associations between free testosterone and ascending aortic distensibility (AAD), during a follow-up of 10 years. In fact, at baseline, women with free testosterone at the highest tertile versus lowest tertiles had lower values of AAD, adjusting for risk factors (b-coefficient = −0.10, 95% CI: −0.19 to −0.01). This study 91 could not demonstrate cross-sectional or longitudinal associations between free testosterone in tertiles and PWV. In an analysis of 411 consecutive middle-aged women, levels of FAI were directly associated with arterial stiffness in a study of non-causal design. 86 More specifically, FAI was shown to directly predict PWV (beta = 0.149, p value = 0.014), stiffness index (beta = 0.154, p value = 0.022), combined measure of local and aortic arterial elastic properties (beta = 0.193, p value = 0.02). Using structural equation modelling analysis, this study also highlighted that the link between FAI and arterial stiffness is not mediated by FAI. The small cohort study by Georgiopoulos et al. 84 reported that higher values of FAI at baseline are linked with a greater risk for transition into the category of abnormal PWV during a follow-up period of 29 months, taking cardiovascular risk factors into account. The results of longitudinal analyses seem to be supportive of a direct link between higher serum levels of testosterone and arterial stiffness in middle-aged women.
The possible link between serum levels of androgens and subclinical atherosclerosis was assessed by two cohort studies. Kische et al. 92 provided cross-sectional and longitudinal associations between circulating androgens and carotid IMT as well as incident atherosclerotic plaque burden in a total of 2140 individuals aged on average 60.8 years and retrieved from the Study of Health in Pomerania. They showed a cross-sectional association between higher total testosterone and IMT, as well as a significant negative association between SHBG and incident plaque burden. 92 However, they could not confirm any consistent longitudinal association between total testosterone and subclinical CVD. 92 The earlier sub-analysis of the CARDIA (Coronary Artery Risk Development in Young Adults) study evaluated 1629 women who were followed up for 20 years. 89 Coronary IMT measures within the highest quartile were associated inversely with levels of SHBG (Q4 levels, OR = 0.56, 95% CI: 0.37–0.84, p value = 0.005), while no associations were observed with total and free testosterone. Overall, longitudinal studies suggest that higher SHBG levels protect from the progression of carotid wall thickening, while there is no convincing evidence on the effect of total or free testosterone.
The possible interrelation between serum levels of circulating androgens and coronary artery calcium (CAC) has been investigated by three studies. The MESA study 90 described a lack of cross-sectional association between prevalent CAC and serum androgen. The longitudinal analysis 90 showed that the CAC-progression ratio increased in parallel with higher levels of free testosterone (2.16, 95% CI: 1.01–1.56), and the CAC-progression risk decreased with higher levels of SHBG (0.80, 95% CI: 0.64–0.99). Two sub-analyses of the CARDIA study tried to identify hormonal predictors of CAC.88,89 In a sample of 1629 women who were followed up for 20 years, 89 the incidence of CAC was inversely associated with values of SHBG above rather than below the median (adjusted OR = 0.59, 95% CI: 0.40–0.86, p value = 0.008), but not with free or total testosterone. More recently, in another subanalysis of the CARDIA study, 88 the incidence of CAC was found to be associated with values of the cortisol to free testosterone ratio within the highest tertile versus the lowest tertile (OR = 3.45, 95% CI: 1.18–10.06). Overall, evidence retrieved from longitudinal studies shows a protective effect of higher SHBG levels regarding the CAC progression. Evidence on the effect of testosterone levels with regards to CAC risk and progression is largely inconsistent.
Clinical CVD
Data on cohort studies assessing the possible associations between endogenous androgens and prevalent or incident rates of CVD is presented in Table 4.
Cohort studies exploring associations between endogenous androgens and cardiovascular events as well as all-cause mortality.
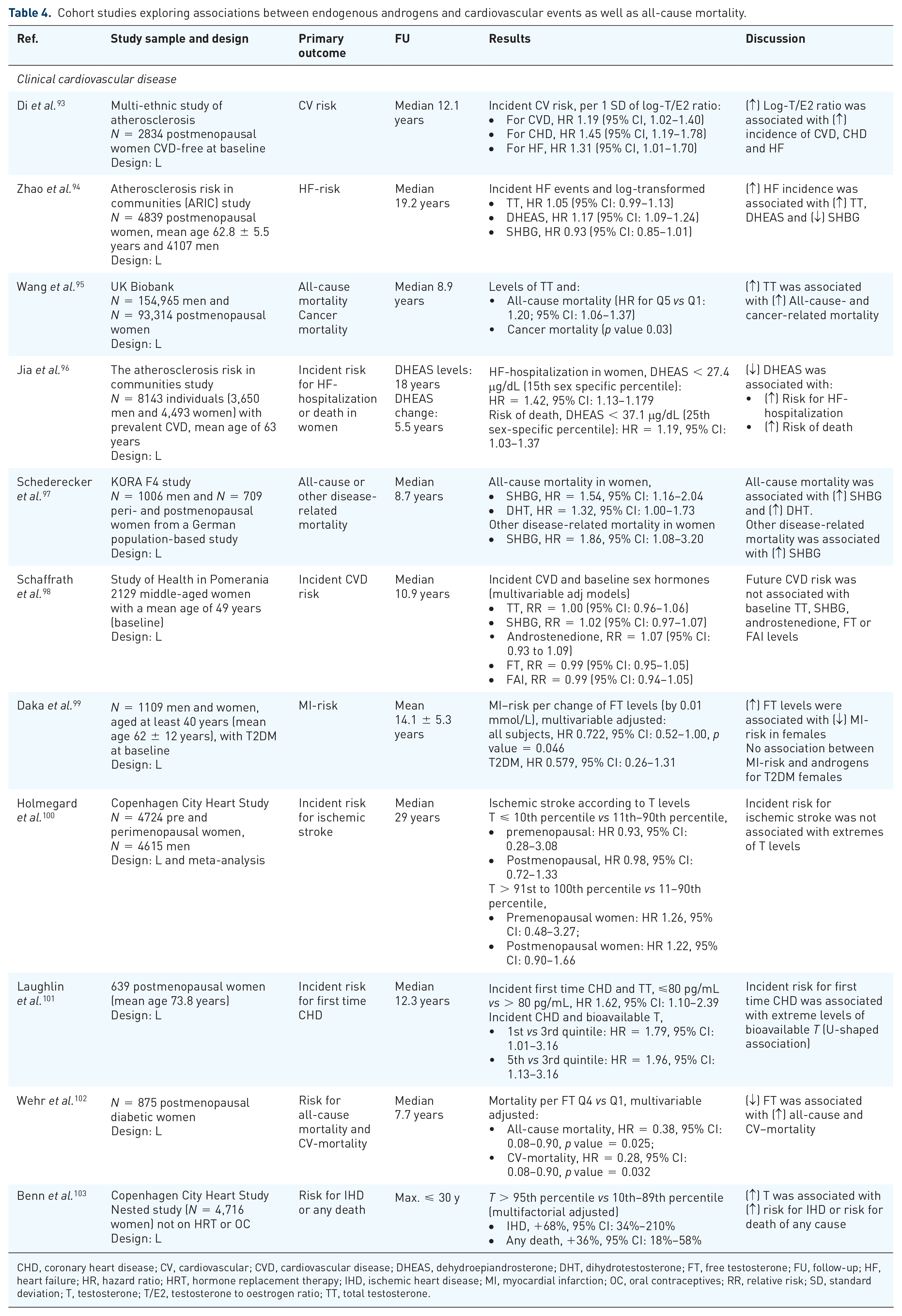
CHD, coronary heart disease; CV, cardiovascular; CVD, cardiovascular disease; DHEAS, dehydroepiandrosterone; DHT, dihydrotestosterone; FT, free testosterone; FU, follow-up; HF, heart failure; HR, hazard ratio; HRT, hormone replacement therapy; IHD, ischemic heart disease; MI, myocardial infarction; OC, oral contraceptives; RR, relative risk; SD, standard deviation; T, testosterone; T/E2, testosterone to oestrogen ratio; TT, total testosterone.
Five of the available cohort studies support a direct association between a more androgenic profile and cardiovascular and/or cerebrovascular events. The possible link between serum levels of androgens and clinical CVD has been investigated in 2834 postmenopausal women from the MESA. 93 This study showed that the risk for future events associated with more pronounced androgenicity, reflected by the testosterone to oestrogen ratio as estimated at 12 years of follow-up (incident CVD, HR = 1.19, 95% CI: 1.02–1.40; incident CHD, HR = 1.45, 95% CI: 1.19–1.78; incident HF, HR = 1.31, 95% CI: 1.01–1.70). Similar results were presented by the Atherosclerosis Risk in Communities study (ARIC), 94 which showed that the risk for incident HF was associated with higher levels of DHEAS (HR = 1.17, 95% CI: 1.09–1.24), but not with total testosterone (HR = 1.05, 95% CI: 0.99–1.13) or SHBG (HR = 0.93, 95% CI: 0.85–1.01). The UK biobank analysis 95 provided valuable insight into possible hormonal predictors of all-cause mortality and described in a total of 93,314 postmenopausal women that higher all-cause mortality was associated with higher total testosterone (HR for Q5 vs Q1: 1.20; 95% CI: 1.06–1.37). The German population-based study (KORA F4) evaluated 709 peri- and postmenopausal women as well as 1006 men for up to 8.7 years, aiming to describe possible links between serum androgens and mortality. 97 This study concluded that female-specific all-cause mortality was associated with higher levels of SHBG and dihydrotestosterone (SHBG, HR = 1.54, 95% CI: 1.16–2.04; dihydrotestosterone, HR = 1.32, 95% CI: 1.00–1.73), while another disease-related mortality was associated with lower androgenicity (SHBG, HR = 1.86, 95% CI: 1.08–3.20). On the contrary, a nested prospective study of 4716 women not on oral contraceptives or hormone replacement therapy 103 reported that higher testosterone was associated with increased risk of ischemic heart disease or death.
The observed positive association between higher androgen levels and increased risk of CVD may be mediated by insulin resistance. Menopause and ageing are associated with lower levels of SHBG, which is a marker of both insulin resistance and of increased androgenicity104,105
On the contrary, three studies support an inverse association between levels of circulating androgens and the risk for cardiovascular and/or cerebrovascular events. One more analysis of the ARIC study 96 demonstrated that the possible link between DHEAS and risk for hospitalizations due to heart failure or the risk of death was evaluated in a total of 8143 individuals with prevalent CVD. The incident risk for hospitalization due to HF was higher in women with DHEAS < 15th sex-specific percentile (HR = 1.42, 95% CI: 1.13–1.17). 96 Similarly, the risk of death was higher for women with levels of DHEAS < 25th sex-specific percentile (HR = 1.19, 95% CI: 1.03–1.37). 96 Investigating a sample of 1109 men and women aged at least 40 years, one more study reported a borderline significant association between low androgenicity and increasing risk of acute myocardial infarction, at least in women with type 2 diabetes, 99 adjusting for cardiovascular risk factors. Similarly, a study of 875 postmenopausal women followed up for up to 7.7 years reported an inverse association between high free testosterone and low cardiovascular and all-cause mortality in postmenopausal women diagnosed with diabetes mellitus. 102
Different results were presented in three more cohort studies. Interestingly, an earlier study 101 described a U-shaped association between levels of testosterone and coronary heart disease in a sample of 639 postmenopausal women, followed up for up to 12.3 years. In conclusion, data retrieved from cohort studies seems to be supportive of a direct link between more pronounced androgenicity and the risk for clinically evident CVD. Finally, two cohort studies of 2129 middle-aged women 98 and 4724 pre- and perimenopausal women 100 could not find longitudinal associations between circulating androgens and clinically evident CVD.
Exogenous androgens and CVD
Trials of testosterone replacement in middle-aged women
Data retrieved from trials of testosterone replacement in middle-aged women are presented in Table 5. In an early trial, 106 administration of a testosterone implant of 50 mg for a total of 6 weeks in postmenopausal women already on HRT described that testosterone administration resulted in improvement of endothelium-dependent and independent vasodilation (increase in FMD, 6.4% ± 0.7% to 9.1% ± 1.1, p value = 0.003; GTN induced, 14.9% ± 0.9 to 17.8 ± 1.2, p value = 0.03). One more trial 107 estimated the impact of intramuscular hormone replacement with 2–5 mg of estradiol esters and 50–100 mg testosterone esters in postmenopausal women compared to never users of HRT. The study 107 concluded that combined estradiol and testosterone treatment for at least 1 year was associated with a significantly higher risk of severe aortic calcification (OR = 3.1, 95% CI: 1.1–8.5). Penotti et al. 108 described the effect on arterial resistance of treatment with testosterone undecanoate 40 mg/day in combination with sequential HRT for 8 months as opposed to HRT only in a small sample of postmenopausal women. The combined oestrogen and testosterone administration resulted in an increase in the pulsatility index of the middle cerebral artery as opposed to baseline. The effect of the oestrogen-testosterone combination was also significantly higher than the equivalent pulsatility index estimated in HRT-only users. One more intervention trial 69 investigated the possible effect of DHEA administration of 100 mg per day in 36 healthy postmenopausal women compared to placebo and concluded that DHEA treatment was related to significant improvement in FMD values (intervention group, 8.4% ± 0.7% to 14.5%±1.1, p value < 0.05; placebo, 10.8% ± 1.1% to 10.9% ± 0.6, p value = NS), but had no effect on the GTN-induced vasodilation.
Studies exploring the possible associations between exogenous androgens and vascular disease.

CI, confidence interval; CV, cardiovascular; DHEA(S), dehydroepiandrosterone (sulphate); E + T, oestrogen and testosterone; FMD, flow-mediated dilation; FU, follow-up; GTN, glyceryl trinitrate; HRT, hormone replacement therapy; IM, intramuscular; m, months; MPA, medoxyprogesterone acetate; NS, not significant; OR, odds ratio; PI, pulsatility index; T, testosterone.
Data on transgender people
Insight into the possible cardiovascular implications of testosterone treatment can be gained from studies on female-to-male transgender. The behavioural risk factor surveillance system (BRSS, 2014–2017) 109 evaluated the risk of myocardial infarction in a large sample of transgender men and women as well as cisgender women (transgender men and cisgender women 410,828; transgender women 1788, cisgender men 306,046). This analysis 109 showed that female-to-male transgenders had higher rates of myocardial infarction compared with cisgender men (OR 2.53, 95% CI: 1.14–5.63, p value = 0.02) and also compared with cisgender women (OR 4.9, 95% CI: 2.21–10.90, p value < 0.01). Additional data retrieved from the BRFSS analysis 110 showed that the elevated crude risk for myocardial infarction observed in transgender men as opposed to cisgender women is largely affected by confounders and highlighted differences in health behaviours between female-to-male transgenders and cisgender women. Transgenders are more frequently overweight (female-to-male vs cisgender women, adjusted odds ratio 1.54, 95% CI: 1.07–2.24), smokers (female-to-male vs cisgender women, OR 1.64, 95% CI: 1.17–2.31), with likely lower educational attainment (combined rates, 30.5% high school graduates; 13.3% never graduated from high-school) and likely to face challenges on employment (5.8% unemployed and 5.8% not able to work). 110 A recent study evaluated the incidence of cardiovascular events in a large transgender cohort, 111 consisting of 2517 male-to-female and 1358 female-to-male (median age of 30 years and 23 years, respectively). Results in male-to-female transgender showed that the standardized incident ratios (SIR) for the development of acute cardiovascular events were higher for stroke, myocardial infarction, and venous thromboembolism (VTE) as opposed to the equivalent risk for cis women (standardized incidence ratio, stroke: 2.42, 95% CI: 1.65–3.42; MI, 2.64, 95% CI: 1.81–3.72; VTE, 5.52, 95% CI: 4.36–6.90). 111 A recent observational analysis of 114 transgender and 964 cisgender individuals 112 described a higher risk of VTE in male-to-female transgenders compared to cis women (adjusted OR 3.94, 95% CI: 1.24–12.51), but comparable rates of any CVD condition in transgender individuals vs cis women or cis men. A prospective cohort study assessed 2842 male-to-female transgenders (followed up for 4 years) and 2118 female-to-male transgenders (followed up for 3.6 years) and compared the cohort with a matched group of 48,686 cisgender men and 48,775 cisgender women. 113 The results showed 113 that the rates of VTE, ischemic stroke or myocardial infarction did not differ between the female-to-male transgenders and the cisgender women (adjusted HR 1.1, 95% CI: 0.6–2.1; 1.3, 95% CI: 0.7–2.5; 1.3, 95% CI: 0.5–3.9; for VTE, stroke and MI, respectively). An earlier meta-analysis of 16 studies, including 1471 male-to-female and 652 female-to-male individuals, showed a low incidence of cardiovascular events in the female-to-male transgender group (death, two cases; myocardial infarction, two cases; VTE, two cases; stroke, no case). 114
Data on the cardiovascular implications of female-to-male transgenders remains limited, and a detailed review of studies on the topic is outside the scope of this study, mainly due to the methodological differences between women on gender-affirming hormone treatment and the peri- or postmenopausal population. The majority of studies on female-to-male transgenders were observational and usually consisted of a small sample size of younger individuals, while the effect of health behaviour possibly modifying the overall cardiovascular risk was not always accounted for. A recent meta-analysis 115 consisting of 29 studies with moderate risk of bias reported that sex hormone treatment in transgender men is usually accompanied by higher rates of dyslipidaemia (at ⩾24 months, mean difference, low-density lipoprotein (LDL) cholesterol, +17.8 mg/dL; 95% CI: 3.5–32.1; serum triglycerides, +21.4 mg/dL, 95% CI: 0.14–42.6). These observations were also supported by a systematic review of 13 studies, 116 which described the metabolic effects of parenteral testosterone treatment (undecanoate 1000 mg per 12 weeks) for up to 60 months in transgender men. The same study 116 reported that transgender men on testosterone treatment have consistently higher levels of LDL-cholesterol and also increased body mass index (from 1.3% to 11.4%). In any case, data on the effect of testosterone treatment in transgender individuals should not be considered directly comparable with the effect of lower-dose testosterone replacement in middle-aged women, considering the difference in age, health behavior, and overall metabolic burden.
Conclusion
Data on the possible effect of testosterone on cardiovascular cells and tissues remain contradictory. Preclinical studies highlight a possibly beneficial effect on vascular cell lines, which fails to be translated into an actual clinical benefit. The results of a recent study on cardiomyocytes 58 and human umbilical vein endothelial cells 59 have revealed that the optimal oestrogen-to-testosterone ratio is ideal, as sex hormones at this level have been found to reduce apoptosis of cardiac myocytes and improve the overall metabolism. Even though the idea of an ‘optimal’ oestrogen-to-testosterone ratio sounds promising, this balance is extremely difficult to be achieved in clinical praxis due to the existence of other counterregulatory factors, which are not present at the preclinical setting. In this context, factors like weight gain and adipose tissue, 117 metabolic syndrome, or even insulin resistance 104 and diabetes mellitus are pathophysiologically interrelated with the oestrogen-to-testosterone ratio and, therefore, likely to modify the effect of sex hormones on the cardiovascular system.
This narrative review summarizes the interplay between the hormonal alterations of the menopausal transition and the cardiovascular system, focusing mainly on androgens. Data on the possible effect of testosterone on cardiovascular cells and tissues remain contradictory. Also, preclinical studies highlight a possible beneficial effect on vascular cell lines, which fails to be translated into an actual clinical benefit. Clinical studies exploring the associations between endogenous androgens and subclinical vascular disease or clinically evident cardiovascular events show an increased risk in women with more pronounced androgenicity, though a possible mediatory effect of insulin resistance cannot be ruled out. On the contrary, several clinical studies describe neutral or inconclusive participation of total and/or free testosterone on cardiovascular endpoints. The idea of an ‘optimal’ oestrogen-to-testosterone ratio sounds promising. Yet this balance is extremely difficult to be achieved in clinical praxis due to the existence of other counterregulatory factors, which are not present at the preclinical setting. In this context, factors like weight gain and adipose tissue, metabolic syndrome or even insulin resistance and diabetes mellitus are pathophysiologically interrelated with the oestrogen-to-testosterone ratio and are, therefore, likely to modify the effect of sex hormones on the cardiovascular system. Evidence on the effect of testosterone treatment remains insufficient to draw firm conclusions. In any case, cardiovascular risk stratification should be incorporated in the management of postmenopausal women, especially when indications for testosterone treatment are present, to minimize the possible future cardiovascular risk.