
Abstract
Patients with type 2 diabetes mellitus often require multifactorial pharmacological treatment due to different comorbidities. An increasing number of concomitantly taken medications elevate the risk of the patient experiencing adverse drug effects or drug interactions. Drug interactions can be divided into pharmacokinetic and pharmacodynamic interactions affecting cytochrome (CYP) enzymes, absorption properties, transporter activities and receptor affinities. Furthermore, nutrition, herbal supplements, patient’s age and gender are of clinical importance. Relevant drug interactions are predominantly related to sulfonylureas, thiazolidinediones and glinides. Although metformin has a very low interaction potential, caution is advised when drugs that impair renal function are used concomitantly. With the exception of saxagliptin, dipeptidyl peptidase-4 (DPP-4) inhibitors also show a low interaction potential, but all drugs affecting the drug transporter P-glycoprotein should be used with caution. Incretin mimetics and sodium–glucose cotransporter-2 (SGLT-2) inhibitors comprise a very low interaction potential and are therefore recommended as an ideal combination partner from the clinical–pharmacologic point of view.
Keywords
Introduction
Patients with type 2 diabetes mellitus (T2DM) often do not suffer solely from symptoms of increased blood glucose levels. In the majority of cases, several comorbidities are present with the need of additional pharmacological treatment. Concomitant diseases such as hypertension and high blood lipids can lead to both microvascular and macrovascular complications [Cornier et al. 2008]. Moreover, central nervous disorders such as depression are increased in patients with T2DM compared with the general population [Anderson et al. 2001]. Multifactorial pharmacotherapy significantly reduces the risk of cardiovascular (CV) mortality [Gaede et al. 2008], but an increasing number of medications taken by the patients leads to a higher risk of adverse drug effects and interactions [Freeman and Gross, 2012; Amin and Suksomboon, 2014; Rehman et al. 2015; Valencia and Florez, 2014; Peron et al. 2015]. Applying a multifactorial pharmacotherapy approach, it is important to consider cytochrome P-450 (CYP) enzyme interactions [De Wildt et al. 1999; Dresser et al. 2000], altered absorption properties [Fleisher et al. 1999] and transporter activities [Lin and Yamazaki, 2003]. Furthermore, nutrition [Fleisher et al. 1999], herbal supplements [Rehman et al. 2015] and other parameters such as patient’s age and gender [Meibohm et al. 2002; Mangoni and Jackson, 2004] are of importance when the drug interaction risk of a pharmacological therapy is assessed.
This article provides a short review of the pharmacokinetic and pharmacodynamic properties of antidiabetic drugs and their clinically relevant interactions with common medications which are frequently used to treat diabetic comorbidities. Literature searches included PubMed and Scopus databases using the Medical Subject Headings (MeSH) terms ‘drug interactions’, ‘diabetes mellitus, type 2/drug therapy’, ‘humans’ and ‘hypoglycemic agents/adverse effects’. Each abstract was studied and the corresponding papers were obtained if considered relevant. Additional studies were identified by citation checking of the reference lists of the studies identified initially.
Types of drug interactions
A drug interaction is defined either as increase or decrease of a medical diagnostic or therapeutic effect of a specific drug caused by another substance, which may be another drug, plant or a dietary supplement. Mechanisms of drug interactions can be divided into two categories: (1) pharmacokinetic interactions, which influence absorption, distribution, metabolism or excretion of a drug (ADME rule) and thus lead to increased or reduced plasma levels of a drug; and (2) pharmacodynamic interactions, which alter pharmacologic efficacy of a drug while drug plasma levels remain unaltered [Rang and Dale, 2012]. Different targets for drug interactions are shown in Figure 1.

Potential mechanisms of drug interactions.
Pharmacokinetic interactions
In the case of pharmacokinetic drug–drug interactions, at least one drug affects the metabolic pathway of the other concomitantly taken drug. The interaction results in either increased or reduced plasma levels of one or both interacting medications compared with plasma levels when the drugs are taken separately.
A frequent mechanism of pharmacokinetic interactions is inhibition or induction of degrading liver enzymes [Dresser et al. 2000]. Even if, in principle, every drug metabolizing enzyme can be the cause for a drug–drug interaction, most interactions are based on oxidative metabolism by the CYP enzyme system [De Wildt et al. 1999], or on an interaction with the drug transporter P-glycoprotein [Lin and Yamazaki, 2003]. In this context, genetic differences are also of clinical importance, as drugs being metabolized by CYP enzymes can vary substantially in their elimination rate depending on the genetically determined enzyme activity [Holstein et al. 2012]. Further-more, altered plasma protein binding (only the free fraction of a drug in plasma is pharmacologically active, displacement from plasma protein binding can increase the active proportion of a drug), absorption and excretion (e.g. by influencing tubular reabsorption) can be mechanisms of pharmacokinetic drug interactions [Fleisher et al. 1999]. For instance, altered gastric pH or the formation of insoluble complexes inside the gastrointestinal tract can result in altered absorption rates. In this regard, food intake and nutritional supplements can play a relevant role by causing significant differences in the plasma concentration of several drugs. A clinical example, relevant for treatment of diabetic patients, is the reduced and slightly delayed metformin absorption rate when drug intake takes place simultaneously with food ingestion [Scheen, 1996; Fleisher et al. 1999]. Moreover, there are gender-specific pharmacokinetic and pharmacodynamic differences to mention [Franconi et al. 2007], even if these differences seem to be of rather minor clinical importance and do not appear to play a clinically relevant role in the treatment of diabetes mellitus [Meibohm et al. 2002].
Herbal drugs represent a complex problem when taken concomitantly with a pharmacological treatment. In the majority of cases insufficient information about the intake of herbal drugs is available for the respective physician, because herbal drug preparations are available over-the-counter. However, these preparations often consist of a complex mixture of bioactive substances, which can interact with pharmacological medications in a different and unpredictable manner. An example for an often used herbal preparation which presents a high interaction potential with several commonly used drugs is St John’s wort due to induction of various hepatic CYP enzymes (CYP3A4, 1A2, 2D6, 2E1) and P-glycoprotein [Dürr et al. 2000; Gurley et al. 2002; Mills et al. 2004]. Thus, St John’s wort affects the disposition of Sulfonylureas and probably Thiazolidinediones, Meglitinides, Sitagliptin, and Saxagliptin.[Xu et al. 2008, Rehman et al. 2014]. Similarly, aloe vera, ginseng, Andrographis paniculata, karela, lycium, and herbs with isoflavones or levocarnitine as ingredients might affect antidiabetic drug metabolism [Rehman et al. 2015]. Patients receiving one of these herbs in combination with antidiabetic drugs metabolized by these enzymes should be closely monitored (Table 1).
Relevant herb-drug interactions with commonly prescribed antidiabetic drugs [Holstein et al., 2012, Rehman et al., 2014].

As patients with T2DM often need to be permanently treated with several different drugs, the uncontrolled intake of herbal preparations represents an important risk factor, in particular in the diabetic patient population [Rehman et al. 2015]. Diabetic patients should be strictly advised not to take any herbal drug preparation concomitantly with antidiabetic medication, without previously consulting their diabetologist.
The age of T2DM patients is often advanced and thus represents a frequent reason for additional complications related to pharmacotherapy [Ng et al. 2014]. In the general population, the number of prescribed drugs is rising with advanced age, with every third over 65-year-old taking five or more different prescribed drugs per quarter [Qato et al. 2008]. In the population of T2DM patients, polypharmacy is even more frequent [Kirkman et al. 2012]. Adverse drug effects such as cognitive impairment, falls resulting from dizziness, weight change and concomitant heart disease are seen more often in patients with polypharmacy. In general, the more drugs that are taken, the more often a clinically relevant drug interaction is to be expected [Delafuente, 2003; Peron et al. 2015]. Moreover, heterogeneity of aged diabetes patients is particularly high and requires a more individual approach to drug therapy [Valencia and Florez, 2014]. With increasing age, liver and renal capacity decreases and so does the ability to metabolize and to eliminate drugs. In particular, the decrease in renal function with increasing age is of major clinical importance due to the frequent need of dose adjustments according to current glomerular filtration rate. Moreover, renal impairment is underdiagnosed in the elderly because, due to lower muscle mass with increasing age, plasma creatinine is not necessarily increased [Breton et al. 2011]. Falls are a frequent problem in advanced age and, in patients with diabetes, often caused by hypoglycemia. The risk of hypoglycemia and similarly the risk of falls depends mainly on the selected antidiabetic agent. The risk is particularly high in case of insulin use, while newer insulin analogs are somewhat safer due to their improved kinetic profile [Bolli et al. 1999]. The risk for hypoglycemia is almost as high when SUs are prescribed in the elderly [Matyka et al. 1997].
Pharmacodynamic interactions
Pharmacodynamic interactions affect either the pharmacologic efficacy or the magnitude of side effects of a drug without affecting its plasma levels. The interaction takes place when concomitantly taken drugs act on the same receptor, or when the receptor binding affinity or its action is altered. The quality of the interaction can be synergistic, additive or antagonistic, or can result in a general increase of adverse events. An example of a desired pharmacodynamic interaction is the additive blood glucose lowering effect by the combination of two or more antidiabetic agents [American Diabetes Association, 2013]. Unfortunately, antidiabetic combination therapy often increases the risk for hypoglycemia, which also can be considered a pharmacodynamic interaction. The risk is substantially increased, especially when an SU is a component of the antidiabetic therapy [Bennett et al. 2011]. The clinically most relevant pharmacodynamic interactions of antidiabetic combination therapy are weight gain, fluid retention and hypoglycemia, which are most frequent when SUs, thiazolidinediones (TZDs) or insulins are used [Freeman and Gross, 2012]. Pharmacodynamic interactions with concomitant medications are of clinical importance in patients with T2DM when drugs are used that affect glucose metabolism. Of particular clinical importance is a concomitant therapy with beta blockers (which may enhance the hypoglycemic effect of SUs), thiazides (which may impair insulin sensitivity, increase insulin resistance, increase basal insulin concentrations and increase plasma glucose concentrations), niacin (which may increase blood glucose levels) or systemic glucocorticoids (which increase blood glucose levels) [Rodbard et al. 2009]. Moreover, there are many pharmacodynamic interactions of herbs that result in a rise or fall in blood glucose levels, thereby disturbing the control of diabetes. Examples are Coccinia indica, ginseng, Gymnema sylvestre, aloe vera, agrimony, alfalfa, cocoa, coffee, elder, fenugreek, flaxseed, holy basil, and herbs with glucosamines as ingredients [Yeh et al. 2003; Patel et al. 2012; Rehman et al. 2015]. Patients receiving one of these herbs in combination with antidiabetic drugs should be closely monitored.
Specific drug–drug interactions with antidiabetic agents
Most common clinically relevant drug–drug interactions with antidiabetic medications occur with SUs, metformin and TZDs (pioglitazone, rosiglitazone). The relevant interactions and the subsequent management are summarized in Table 2. Drug–drug interactions are of special concern when new drugs are given or the dosage of a respective medication is adjusted. This is often the case when the clinical condition temporarily requires anti-infective therapy. An example is the elevated risk of hypoglycemia when patients on treatment with an SU are temporarily treated with clarithromycin, which acts as a potent CYP3A4 and P-glycoprotein inhibitor [Bussing and Gende, 2002]. In the following paragraphs antidiabetic drugs and their relevant interactions are summarized.
Substance-specific drug–drug interactions with antidiabetic medication in clinical routine.
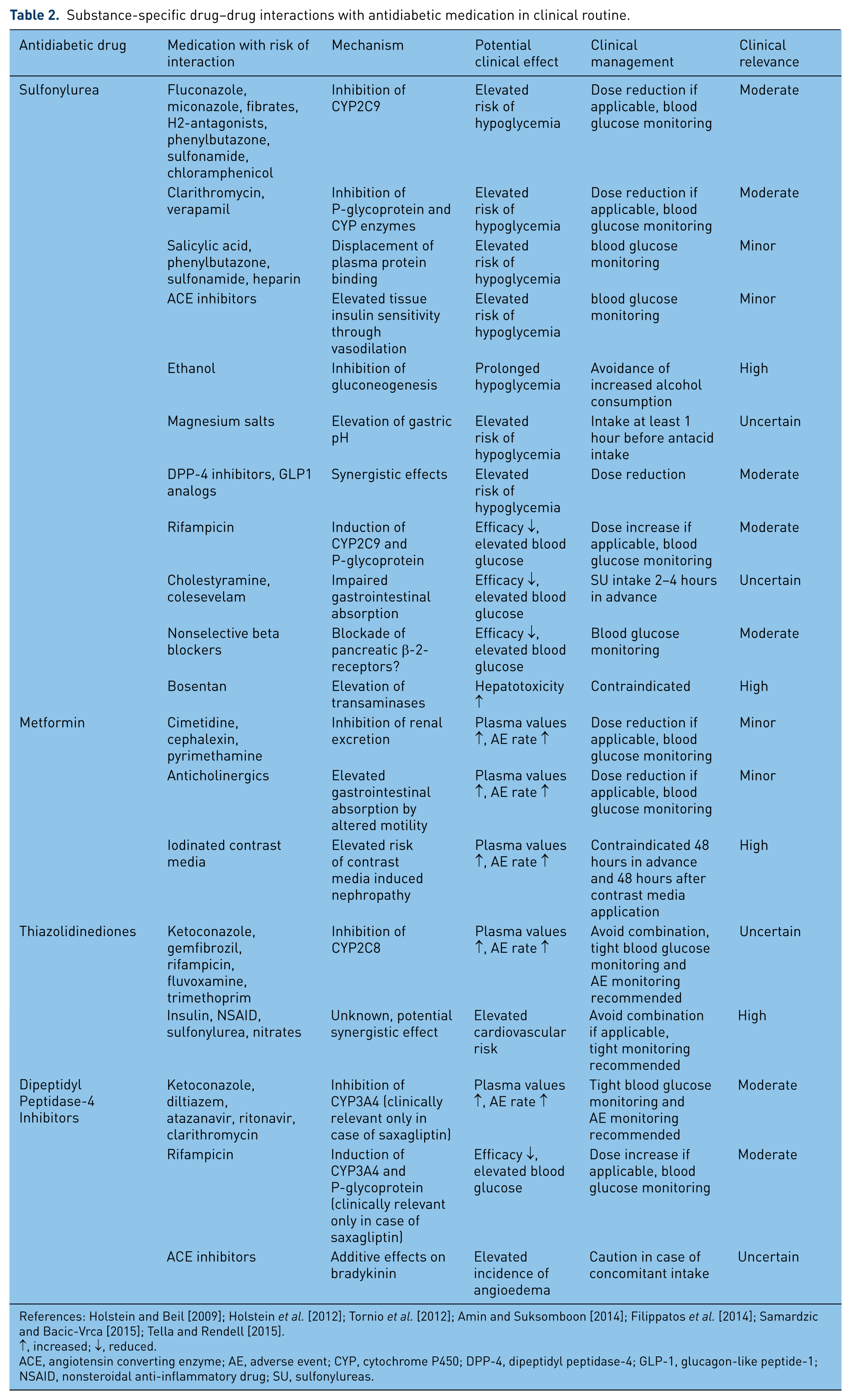
References: Holstein and Beil [2009]; Holstein et al. [2012]; Tornio et al. [2012]; Amin and Suksomboon [2014]; Filippatos et al. [2014]; Samardzic and Bacic-Vrca [2015]; Tella and Rendell [2015].
↑, increased; ↓, reduced.
ACE, angiotensin converting enzyme; AE, adverse event; CYP, cytochrome P450; DPP-4, dipeptidyl peptidase-4; GLP-1, glucagon-like peptide-1; NSAID, nonsteroidal anti-inflammatory drug; SU, sulfonylureas.
Drug–drug interactions with common comedication in diabetic patients
Many patients with diabetes and increased CV risk are treated with statins, which are predominantly metabolized by CYP3A4 (e.g. simvastatin, lovastatin, atorvastatin and fluvastatin). When taken concomitantly with glibenclamide (= glyburide), statins have the potential to increase the maximum plasma concentration (Cmax) and the area under the curve (AUC) of glibenclamide by up to 20%. However, the clinical importance of this interaction remains unclear [Holstein et al. 2012].
Angiotensin converting enzyme (ACE) inhibitors can increase the tissue sensitivity for insulin and thus cause slight vasodilation which, in principle, may increase the risk of hypoglycemia. This interaction is also of questionable clinical relevance (Table 2).
Patients with T2DM are often comedicated with vitamin K antagonists such as warfarin due to CV comorbidity. When warfarin (or phenprocoumon) is taken concomitantly with an SU, an elevated risk for hypoglycemia is stated in several databases. However, little is to be found in the literature about the proposed interaction. Only a single case report describes an interaction between warfarin and glibenclamide in a warfarin-maintained patient whose International Normalized Ratio (INR) became elevated during glibenclamide treatment [Armstrong et al. 1991]. Thus, the mechanism of the proposed interaction is still unclear. Displacement from its plasma protein binding sites may be a possible mechanism for the proposed interaction between SUs and coumarins. Due to the lack of clinical studies, the clinical relevance of the proposed interaction is difficult to classify.
Although allopurinol, a drug prescribed for the treatment of gout and hyperuricemia and often used concomitantly in diabetes patients, has a relatively high pharmacological interaction potential, no relevant interactions between allopurinol and any antidiabetic medication are known. Nevertheless, caution and dose adjustment of allopurinol are advised when renal failure is present.
Metformin
Metformin is the only biguanide currently approved for the treatment of T2DM. It is recommended as first-line therapy because of good clinical efficacy and a low incidence of adverse events [American Diabetes Association, 2013]. Metformin is partially absorbed in the small intestine, shows low plasma binding, and is excreted by renal elimination without hepatic metabolism. The elimination rate of the drug is mainly determined by renal function, in which several specific cation transporters are involved [Scheen, 1996]. Thus, all drugs affecting renal function may also reduce the metformin clearance and may thereby increase the adverse event rate of metformin.
In particular, the risk of developing lactic acidosis is still a major concern when treating with metformin today. Lactic acidosis is a rare but potentially life-threatening adverse event of metformin, which occurs especially and much more frequently in patients suffering from moderate to severe renal failure [Rang and Dale, 2012]. Accordingly, metformin is contraindicated 48 hours before and 48 hours after administration of iodinated contrast agents due to the risk of additive contrast-induced renal failure (product information). Limited data are available about provoked lactate acidosis via this proposed mechanism, but all renally eliminated drugs with involvement of the same transporters used by metformin (OCT2, MATE1, MATE2K), or drugs inhibiting these transporters, should be used with caution when metformin is taken concomitantly [Amin and Suksomboon, 2014]. Accordingly, caution is required when patients are concomitantly treated with cimetidine, procainamide, trimethoprim, digoxin, amiloride, quinine, quinidine, ranitidine, vancomycin, cephalexin or pyrimethamine.
Anticholinergics increase the oral bioavailability of metformin by altering gastrointestinal motility. Thus metformin should be used with caution in combination with anticholinergics [Amin and Suksomboon, 2014]. Furthermore, metformin use is associated with anemia and vitamin B12 malabsorption, which may be due to a metformin-mediated effect on small bowel motility and thus decreased vitamin B12 absorption [Liu et al. 2014]. Vitamin B12 deficiency is associated with metformin use in diabetic patients and vitamin supplementation seems to be beneficial even in patients with B12 serum levels within the normal range [Pflipsen et al. 2009]. Significantly decreased serum vitamin B12 levels are a relatively late clinical manifestation of vitamin B12 deficiency. Recommended early markers are holotranscobalamin, also known as active B12, which is the earliest laboratory parameter for B12 deficiency, and methyl malonic acid, which is a functional B12 marker increasing when B12 stores are depleted [Herrmann et al. 2008].
SUs
SUs are acting independently of blood glucose levels by inhibition of adenosine triphosphate-dependent potassium channels of the pancreatic beta cells. Glibenclamide, glimepiride and glipizide are the most frequently used SUs. Treating increased blood sugar levels with an SU implies an increased risk of hypoglycemia due to the glucose-independent mode of action [Rang and Dale, 2012]. SUs have a high bioavailability and are metabolized by CYP2C9 and to a lesser extent by CYP3A4 and CYP2C9 [Holstein and Beil, 2009]. Molecules are largely bound to plasma proteins (95–99%) and are renally eliminated. Only a minor amount of the drug is excreted by feces. High protein binding and hepatic metabolism by CYP enzymes further add to the high drug interaction potential of SUs. Moreover, gastrointestinal absorption depends largely on gastric pH and even small gastric pH changes significantly affect SU bioavailability [Holstein and Beil, 2009; Tornio et al. 2012].
About 100 clinically used drugs including SUs are primarily metabolized by CYP2C9 [Holstein et al. 2012]. Inductors of CYP2C9, such as carbamazepine, phenobarbital, rifampicin, ritonavir and St John’s wort, cause an increased elimination rate and thus result in decreased plasma levels of CYP2C9 substrates like SUs. In contrast, inhibitors of CYP2C9 such as amiodarone, cimetidine, ranitidine, bosentan, trimethoprim, fluconazole, ketoconazole, voriconazole, fluoxetine, fluvaxamine, fluvastatin, leflunomide, metronidazole and noscapine prolong SU degradation [Holstein et al. 2012]. Thereby, when the daily dose of SU is not adjusted accordingly, increased plasma levels can result and clinically dangerous hypoglycemia can occur. This is even more important, when drugs are used temporarily, for example in case of antimicrobial treatment. Many antibiotic drugs affect hepatic enzyme activity and thus substantially increase hypoglycemic risk with SU, and are associated with higher morbidity and increased costs [Parekh et al. 2014]. Thus, interactions due to CYP2C9 inhibition play a crucial role in the treatment with SUs. A Finnish study revealed that a clinically relevant interaction occurred in about 20% of T2DM patients treated with SUs; 75% of the identified interactions were due to concomitant treatment with trimethoprim, metronidazole, or ketoconazole [Tirkkonen et al. 2010].
Moreover, an inhibition of the gastrointestinal drug transporter P-glycoprotein can result in increased plasma concentrations of glibenclamide. This is of particular clinical relevance when verapamil or clarithromycin are taken concomitantly to SUs [Holstein et al. 2012]. Organic anion transporting polypeptides OATP1B1-3 are drug transporters relevant for cellular drug entry and thus mechanism of action of most drugs [Picard et al. 2010]. Recently expression of OATP1B3 was found to be in the islets of Langerhans of human pancreas, and facilitating cellular entry of glibenclamide [Meyer Zu Schwabedissen et al. 2014]. Thus, OATP1B3 likely are affected by drug-drug interactions or by genetic variants, influencing efficacy of SU or Meteglinids. Further studies are needed to determine possible interactions and their clinical relevance. There are many more drug–drug interactions with SUs mentioned in the literature, for which the underlying mechanism is not fully understood. Examples are: elevated liver enzymes with bosentan [van Giersbergen et al. 2002]; increased hypoglycemic potential with concomitant chloramphenicol therapy (product information for glibenclamide); nonsteroidal anti-inflammatory drugs (NSAIDs) or salicylates may increase the hypoglycemic action of SUs [Kubacka et al. 1996]; and there are additive hypoglycemic effects when dipeptidyl peptidase-4 (DPP-4) inhibitors such as sitagliptin and an SU are co-administered [Scheen, 2010]. Another important interaction with SU is that with ethanol, which is why alcohol intake is dangerous for patients taking SUs. Ethanol inhibits hepatic gluconeogenesis and thereby increases the risk of developing hypoglycemia during SU treatment and a disulfiram-like flushing reaction can occur [Lao et al. 1994]. Patients should be encouraged to avoid alcohol intake. If alcohol is consumed nevertheless, intake should not exceed 1 to maximally 3 drinks (10–45 g alcohol) per day and should always be in conjunction with food, to minimize the inherent hypoglycemic effects of ethanol.
Magnesium salts, which are common ingredients of several over-the-counter medicines (antacids), increase the risk of hypoglycemia by increasing the intestinal absorption rate of SUs. Therefore, SUs should be administered at least 1 hour prior to antacid intake. The opposite effect, a decreased intestinal absorption rate, can be expected when cholestyramin is taken concomitantly to SUs [Amin and Suksomboon, 2014].
The mechanism of action of glinides such as nateglinide and repaglinide is similar to SUs. Up to now, a positive effect on long-term survival has not been proven in endpoint trials and thus glinides are of minor clinical importance for the treatment of T2DM patients [American Diabetes Association, 2013]. Unlike SUs, glinides can be prescribed for patients with renal insufficiency. Due to their faster absorption rate and shorter halflife, glinides possess a lower risk of hypoglycemia compared with SUs [Rang and Dale, 2012]. CYP2C8, CYP3A4, uridine diphosphate glucose (UDP) glucuronosyltransferase (UGT) and the organic anion transporter OATP1B1 are involved in the elimination of repaglinide. Relevant interactions exist with cyclosporine and gemfibrozil, which should not be administered concomitantly with repaglinide. As nateglinide and SUs are both metabolized by CYP2C9, potential drug interactions are similar for nateglinide and SUs [Holstein et al. 2012].
TZDs
TZDs, which are also called glitazones, are known as insulin sensitizers. TZDs increase the sensitivity to insulin in monocytes, adipocytes and hepatocytes without affecting pancreatic insulin release. Members of this class are rosiglitazone (approved only in the US), pioglitazone (the only TZD available in Europe) and lobeglitazone (approved for use in Korea only and therefore not discussed in this article). Use of rosiglitazone but not pioglitazone seems to be associated with increased risk of heart failure and myocardial infarction [Kaul et al. 2010]. In 2008 the American Diabetes Association and the European Association for the Study of Diabetes consensus algorithm recommended against the use of rosiglitazone due to concerns that the overall risks exceed its benefits. The European Medicines Agency suspended sales of rosiglitazone in 2010. Fluid and salt retention are common side effects of TZD, implying a negative impact on CV mortality for all members of this drug class. Even if this has not yet been proven in randomized controlled trials, monitoring for signs and symptoms of heart failure is indicated, especially when insulin or an SU are co-administered, which enhances the adverse/toxic effect of TZD. Specifically, the risk of fluid retention, heart failure and hypoglycemia may be increased with this combination (product information). The same might apply to a combination therapy with NSAIDs. In contrast to rosiglitazone, there seems to be no interaction when nitrates and pioglitazone are taken concomitantly [Erdmann et al. 2010]. Furthermore, fracture risk is increased in patients with TZD therapy and seems to be a drug-related adverse event. Pioglitazone was associated with an increased risk of bladder cancer in studies [Rang and Dale, 2012].
Pioglitazone is metabolized mainly by CYP2C8 and to a lesser extent by CYP3A4 [Jaakkola et al. 2006]. Rosiglitazone is predominantly metabolized by CYP2C8 with only minor contribution of CYP2C9 [Cox et al. 2000]. In patients concomitantly treated with TZDs and the CYP inductor rifampicin, an attenuated glucose lowering effect should be considered. When gemfibrozil, a potent CYP2C8 and OATP1B1 inhibitor, is co-administered, the daily dose of a TZD should be halved due to impaired TZD metabolism [Holstein et al. 2012].
DPP-4 inhibitors
DPP-4 inhibitors cause increased incretin levels by selective inhibition of the enzyme DPP-4 and thus increase insulin secretion and inhibit glucagon secretion. The incidence of hypoglycemia is significantly lower with DPP-4 inhibitors compared with SUs. Therefore, the incidence of cardiac events is estimated to be lower, especially in elderly patients [Tella and Rendell, 2015]. Approved members of this class are sitagliptin, saxagliptin, linagliptin, vildagliptin, anagliptin (Japan only), teneligliptin (Japan only) and alogliptin (US only). Vildagliptin was withdrawn from the market in Germany due to a negative benefit risk analysis performed by the federal joint committee (GBA) in July 2014. Linagliptin did not even reach German market authorization due to a negative GBA review, despite its favorable pharmacokinetic properties. Linagliptin is mainly biliary excreted with only minor hepatic metabolism (low interaction potential) and negligible renal elimination, which makes it therapeutically particularly attractive for use in patients with moderate or severe renal impairment. DPP-4 inhibitors appear to have similar glycemic efficacy and result in modest glycated haemoglobin (HbA1c) improvement of about -0.74% [Amori et al. 2007; Scheen et al. 2010]. There are no data on long-term safety, mortality, diabetic complications or health-related quality of life. Recently, DPP-4 inhibitors have been associated with severe joint pain [Tarapues et al. 2013].
Even though the mechanism of action of all DPP-4 inhibitors are similar, different molecules and slightly different pharmacokinetic properties account for different interaction potentials. Common for all DPP-4 inhibitors is an increased incidence of hypoglycemia in combination with an SU compared with the use of an SU alone (product information). It is recommended to consider SU dose reductions in patients receiving both agents and to monitor for the development of hypoglycemia. Nonetheless, with the exception of saxagliptin, which is metabolized via CYP3A4/5, no clinically relevant pharmacokinetic drug–drug interactions are known for the class of DPP4 inhibitors and commonly used concomitant medications in T2DM patients. However, the incidence of angioedema exerted by an ACE inhibitor seems to be higher when the patient is comedicated with a DPP-4 inhibitor, which might be due to an additive effect on bradykinin [Filippatos et al. 2014]. Saxagliptin is metabolized by CYP3A4 and therefore possesses interactions with relevant CYP3A4-inhibitors (e.g. ketoconazole, diltiazem, atazanavir, ritonavir and clarithromycin) or CYP3A4-inductors (e.g. rifampicin) [Filippatos et al. 2014; Tella and Rendell, 2015]. Even if there are no relevant CYP interactions with other DPP-4 inhibitors, they are substrates of P-glycoprotein and organic anion transporters and therefore possess the potential to slightly increase plasma concentrations of digoxin when taken concomitantly [Filippatos et al. 2014]. Importantly, similar to a treatment with metformin and in strong contrast to SU and TDZ treatment, DPP-4 inhibitor treatment does not result in weight gain [Rang and Dale, 2012].
Glucagon-like peptide-1 analogs
Glucagon-like peptide-1 (GLP-1) analogs reproduce the naturally occurring peptide GLP-1 and affect glucose control through several mechanisms, including enhancement of glucose-dependent insulin secretion, slowed gastric emptying, regulation of postprandial glucagon and reduction of food intake [Koliaki and Doupis, 2011]. Exenatide and liraglutide are currently available as analogs of GLP-1 and are approved for the treatment of T2DM. GLP-1 is a peptide hormone of the gut, secreted by the L cells of the intestinal mucosa – it is one of the intestinal incretins. Therefore, members of this drug class are also known as incretin mimetics. Exenatide and liraglutide are administered subcutaneously [Rang and Dale, 2012]. A main therapeutic advantage of this drug class is its glucose-dependent mechanism of action, which significantly reduces drug-induced hypoglycemia. Beyond their blood glucose reducing efficacy, GLP-1 analogs additionally cause relevant reduction of systolic and diastolic blood pressure (by 1–5 mm Hg when compared with other antidiabetic drugs) [Wang et al. 2013], which results in a weight reduction of about -3 kg [Vilsbøll et al. 2012]. Up to now, no clinically relevant drug–drug interactions have been described, either for exenatide or for liraglutide [Samardzic and Bacic-Vrca, 2015]. Only a slight delay of gastric emptying time has been described in the literature, but without evidence for clinically significant influence on the absorption rate of other drugs [Kapitza et al. 2011]. When SUs are taken concomitantly, the SU dose should be halved because, in case of co-administration, the rate of hypoglycemia might be raised due to additive hypoglycemic effects [Amin and Suksomboon, 2014].
Sodium–glucose cotransporter-2 inhibitors
Sodium–glucose cotransporter-2 (SGLT-2) inhibitors are a promising new drug class for the treatment of T2DM [Idris and Connelly, 2009; Chao and Henry, 2010]. SGLT-2 inhibitors reduce blood glucose levels in T2DM patients by inhibiting glucose reabsorption in the proximal tubule, thus subsequently increasing renal glucose excretion. Approved agents for the treatment of T2DM are empagliflozin, dapagliflozin and canagliflozin (the latter is no longer available in Germany). Like incretin mimetics, SGLT-2 inhibitors reduce weight and blood pressure [Clar et al. 2012]. Due to the additional diuretic effect of SGLT-2-inhibitors, dose adjustments may be required in patients on concomitant diuretic therapy [Scheen, 2015]. All SGLT-2 inhibitors exhibit high plasma protein binding. However, clinically significant interactions due to displacement from plasma protein binding have not so far been described. SGLT-2 inhibitors are metabolized in the liver by glucuronidation without involvement of CYP enzymes. With the exception of canagliflozin, which should be given in a higher daily dose when co-administered with UGT inducers such as rifampicin, phenytoin or ritonavir, and which might increase digoxin plasma levels, SGLT-2 inhibitors do not show any clinically relevant interactions, either with other antidiabetic agents or with typically used concomitant medications, which has been proven in numerous clinical interaction studies [Amin and Suksomboon, 2014].
Drugs affecting the efficacy of antidiabetic agents in general
Thiazide diuretics impair antidiabetic drug efficacy by increasing insulin resistance and increasing plasma glucose concentrations due to reduced total body potassium [Salvetti and Ghiadoni, 2006]. A recent study identified thiazide diuretics as the agents with the most frequent drug–drug interaction with antidiabetic medication [Samardzic and Bacic-Vrca, 2015]. Beta blockers are also associated with increased risk of developing T2DM [Sarafidis and Bakris, 2006]. The hyperglycemic effects are probably explained through reduction of pancreatic insulin release [Wicklmayr et al. 1990]. Beta blockers can mask the early warning symptoms of hypoglycemia; however, later studies have shown that these effects may be less prominent with newer beta-1 selective drugs [Shorr et al. 1997]. As mentioned earlier, many antimicrobials present a high interaction risk with antidiabetics, especially with Sulfonylureas [Tirkkonen et al. 2010, Parekh et al. 2014]. Interactions with antimicrobials are clinically most relevant because adverse effects take place most often whenever a drug is added or removed [Patsalos et al. 2003], and antimicrobials are usually taken temporarily. Besides specific pharmacokinetic interactions, which depend on drug metabolism, there are some antibiotics with glucose lowering effects and thus bearing the risk of pharmacodynamic interactions with antidiabetic drugs in general. A clinically relevant example are fluoroquinolones[Chou et al. 2013, Fusco et al. 2013, Parekh et al. 2014]. Quinolones seem to have an insulinotropic effect by increasing the release of insulin via a blockade of ATP-sensitive K+ channels in a dose-dependent manner [Maeda et al. 1996]. Fluoroquinolones like levoflaxacin, ciprofloxacin, or moxifloxacin should be used with caution in patients with diabetes.
Another frequent interaction is with glucocorticoid therapy, which is a particularly common cause of clinically significant drug-induced hyperglycemia. Glucocorticoid therapy has a negative effect on glucose metabolism [Olefsky and Kimmerling, 1976] and is accompanied by substantial weight gain [Hoes et al. 2007].
In contrast, for anabolic steroids such as testosterone, there is evidence for an increase in insulin sensitivity [Kapoor et al. 2006]. The same applies to weight-reducing treatments or procedures [Knowler et al. 2002]. Otherwise, drugs associated with weight gain such as various antipsychotics (chlorpromazine, haloperidole, clozapine, olanzapine, risperidone, quetiapine) have the potential to worsen glucose tolerance and thereby seemingly decrease antidiabetic drug efficacy [Amin and Suksomboon, 2014]. The same applies to hormonal contraceptives, protease inhibitors, niacin and calcineurin inhibitors, which are other drugs with considerable potential to contribute to a deterioration of blood glucose control [Luna and Feinglos, 2001]. Glycemic control may be altered with use of sympathomimetic agents, leading to decreased effects of antidiabetic agents. In contrast, orlistat improves glycemic control and reduction of antidiabetic treatment might be necessary.
More frequent monitoring of blood glucose levels and dose adjustment is recommended in diabetic patients with these comedications. Moreover, ethanol consumption generally should be restricted in patients with diabetes [Franz et al. 2004]. Ethanol can alter glucose tolerance and insulin secretion [Nikkila and Taskinen, 1975] and there is an additive hypoglycemic effect of ethanol in patients receiving antidiabetic agents [Walsh and O’Sullivan, 1974].
Table 3 lists databases known to provide further information on clinically relevant drug–drug interactions.
Drug interaction databases for web-based research.

Conclusion
Most clinically relevant interactions of antidiabetic agents are based on the induction or inhibition of hepatic CYP enzymes. Relevant interactions are predominantly related to SUs, TZDs and glinides. Although metformin possesses a very low interaction potential via metabolizing enzymes, caution is advised for concomitant treatment with drugs impairing renal function. With the exception of saxagliptin, DPP-4 inhibitors also show a very low interaction potential. However, co-administration of drugs affecting the drug transporter P-glycoprotein with a DPP-4 inhibitor might affect plasma levels. Incretin mimetics and SGLT-2 inhibitors as new antidiabetic drug classes possess a very low interaction potential and seem to be ideal combination partners in diabetes therapy from the clinical–pharmacologic point of view.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.