
Abstract
Introduction
Type 2 diabetes is a heterogeneous disease characterized by disturbances in both insulin secretion and peripheral tissue sensitivity to insulin action and expressed in different degrees in particular patients [Borissova et al. 1991].
The different major classes of oral antihyperglycemic agents in current use can be divided into those that increase insulin secretion as sulfonylureas, those that decrease insulin resistance as metformin and thiazolidinediones, and those that modify the rate of glucose entry from the gastrointestinal tract as α-glucosidase inhibitors [Kimmel et al. 2005].
Sulfonylureas stimulate insulin release from pancreatic β-cells by first binding to the high-affinity sulfonylurea receptor (SUR1), which together with the potassium pore-forming inward-rectifier (Kir6.2) subunits make up the pancreatic β-cell ATP-sensitive potassium (KATP) channel. This interaction closes the K+ channel, which inhibits potassium efflux and depolarizes the plasma membrane, leading to an opening of voltage-gated calcium channels. Calcium influx, and a corresponding increase in intracellular calcium levels, causes release of insulin from the β-cells [Distefano and Watanabe, 2010].
Treatment with sulfonylureas is initially successful in type 2 diabetes. However, it is often associated with high secondary failure rate (hemoglobin A1c ≥ 8%) with the highest recommended dose in patients who initially respond to sulfonylurea therapy and consequently switch to other treatment progressively as sulfonylurea fails [Nichols et al. 2007]. The underlying mechanisms are still undefined. Poor compliance with diet, gain in body weight, deterioration of insulin sensitivity, cell exhaustion, conditions requiring the use of a diabetogenic drug, and the presence of anti-islet cell and glutamic acid decarboxylase (GAD) antibodies have been suggested as causative factors. An increased frequency of human leukocyte antigens (HLAB21 and HLADRl) may also play a part. The progressive decline of pancreatic β-cell function has been demonstrated to be the stronger predictor of the insulin-requiring stage in type 2 diabetes [Sesti et al. 2006; Borissova et al. 1991].
Genetic factors are clearly important for the main pathogenic mechanisms leading to common type 2 diabetes, β-cell dysfunction, and impaired insulin action. Among plausible candidate genetic variants that could affect insulin secretion are the E23K polymorphism of the Kir6.2 gene (KCNJ11, on chromosome 11), and polymorphism in insulin receptor substrate-1 (IRS-1) causing a Gly972Arg change (Arg972) [Gloyn et al. 2005; Sigal et al. 1996].
Mice lacking the Kir6.2 gene are characterized by impaired glucose- and tolbutamide-induced insulin secretion. Gain-of-function mutations in Kir6.2 produces permanent neonatal diabetes, whereas loss-of-function polymorphisms in KCNJ11 have been associated with familial hyperinsulinemic hypoglycemia of infancy [Gloyn et al. 2004; Miki et al. 1998; Nestorowicz et al. 1997].
The common E23K polymorphism, in which substitution in codon number 23 of glutamic acid (E) resulted in the codon of lysine (K) amino acid, has been most extensively studied in the classical form of type 2 diabetes, and has been identified and found to be a major diabetogene in different populations [Hani et al. 1997; Lindner et al. 1997; Inoue et al. 1996; Iwasaki et al. 1996]. Some studies demonstrate that the E23K polymorphism increases in intrinsic open probability of KATP channels, with a consequent reduction in ATP sensitivity, and enhanced activation by Mg nucleotides and long-chain acyl-CoAs [Reidel et al. 2003; Schwanstecher et al. 2003].
A study of 525 white patients with type 2 diabetes found a higher frequency of the K allele in patients who failed sulfonylurea therapy compared with those who did not (66.8% versus 58.0%, respectively) [Sesti et al. 2006].
IRS-1 functions as one of the key signaling molecules in the insulin receptor signaling pathway. After autophosphorylation of the insulin receptor, the receptor kinase is activated and phosphorylates IRS-1 and other intracellular substrates. This signaling molecule then acts as a docking protein for multiple Src homology-2 domain (SH2)-containing proteins, including phosphatidylinositol (PI) 3-kinase. Consequently, this leads to activation of several enzymes that may be responsible for mediating insulin action within the target cell. Molecular scanning of the IRS-1 gene in normal individuals and patients with type 2 diabetes has revealed several polymorphisms resulting in amino acid substitutions, the most common of which is a Gly→Arg substitution at codon 972 (Arg972) [Strohmer et al. 2005; Xu et al. 2000].
A meta-analysis of 27 studies including 3408 patients with diabetes and 5419 controls revealed that carriers of the Arg972 IRS-1 variant have an increased risk for type 2 diabetes [Jellema et al. 2003]. Transfection studies have demonstrated that the Gly972Arg change has a functional consequence causing impairment in IRS-1-associated PI 3-kinase activity owing to their defective interaction [Hribal et al. 2000; Porzio et al. 1999]. Several investigations have been performed, usually suggesting that the Arg972 IRS-1 polymorphism could predispose people to type 2 diabetes in certain ethnic groups, such as Finnish, Danish, French and Italian [Bezerra et al. 2004].
Early investigations in pancreatic β-cell lines and human pancreatic islets of Langerhans found that the genotype at the Gly972Arg polymorphism of IRS1 affected the level of insulin secretion in response to sulfonylureas. The clinical impact of this polymorphism was also investigated in white patients with type 2 diabetes who were treated with sulfonylurea agents. In these patients, the genotype frequency of the variant allele (Arg972) was almost twice as high in patients who experienced secondary sulfonylurea failure compared with those with controlled glycemia [Sesti et al. 2004].
To test the intriguing hypothesis that patients with diabetes carrying E23K, and Arg972 polymorphisms are at increased risk for secondary failure to sulfonylurea, the prevalence of the E23K and Arg972 variants were examined in Egyptian patients with type 2 diabetes with and without secondary failure to sulfonylurea.
Material and methods
Patient population
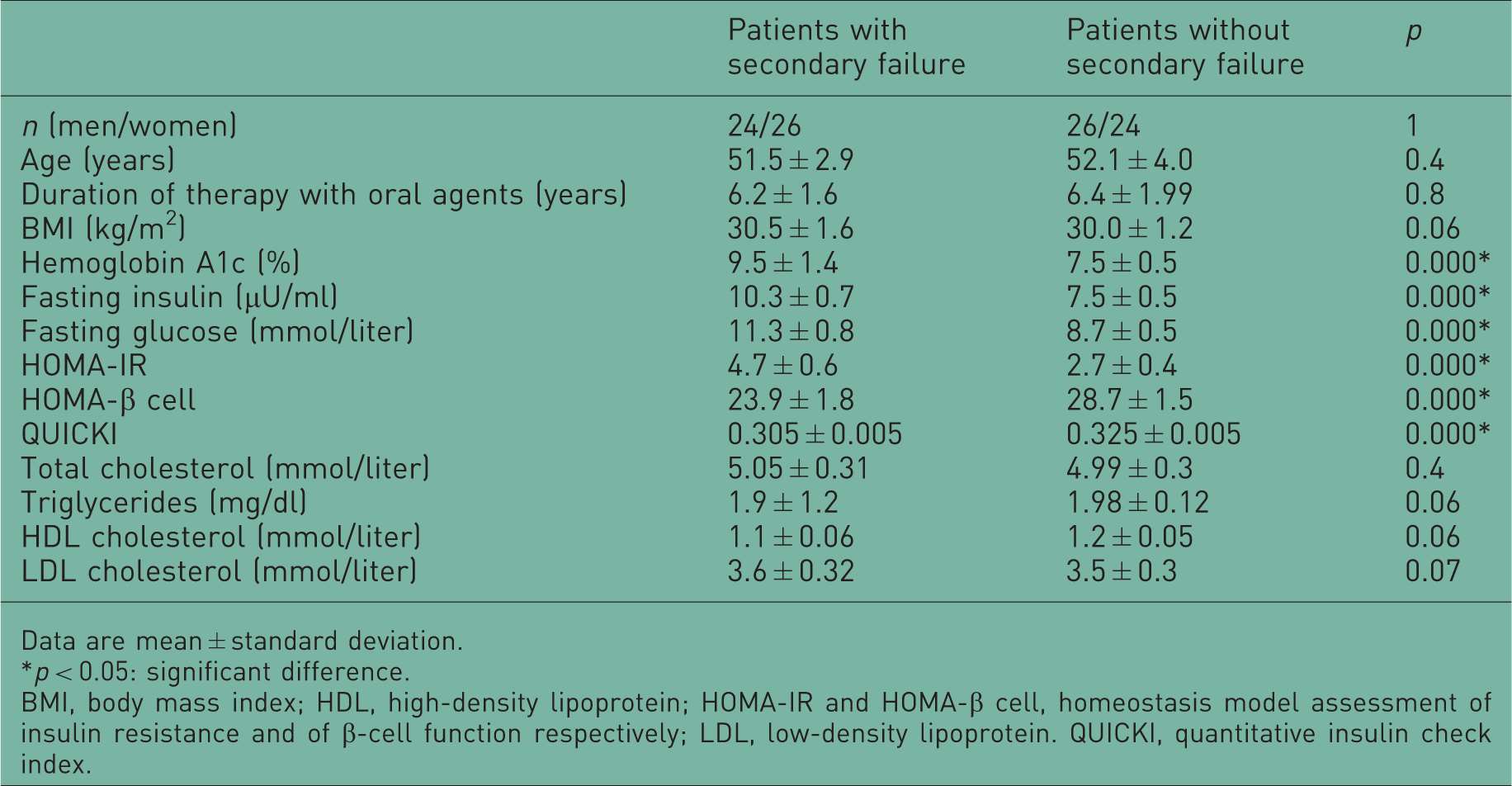
The study was carried out at the Specialized Internal Medicine Hospital, Mansoura University. The study included 100 unrelated Egyptian patients with type 2 diabetes with onset of the condition after the age of 35 years, and absence of the GAD antibody which may be associated with the development of type 1 diabetes. Patients were nonsmokers and their age ranged from 40 to 60 years. Diabetes was diagnosed in accordance with the World Health Organization 1999 criteria. Patients were excluded if they had chronic gastrointestinal diseases associated with malabsorption, chronic pancreatitis, history of any malignant disease, history of alcohol or drug abuse, liver or kidney failure, or clinical problems potentially causing hyperglycemia, including infection, thyroid disease, surgery, and treatments known to modify glucose metabolism. They were divided into two groups (50 patients each): group I included patients with secondary failure to sulfonylurea. Patients with secondary failure are defined as those with uncontrolled hyperglycemia (hemoglobin A1c ≥ 8%) despite sulfonylurea therapy. Group II, the control group, consisted of patients whose condition was controlled with sulfonylurea therapy. The two groups were matched according to mean age, mean body mass index (BMI), duration of therapy and sex distribution. Informed consent was obtained from all patients. This study was approved by the Tanta University ethics committee.
Biochemical assays
Blood samples were taken from patients after overnight fasting in order to measure fasting serum levels of glucose enzymatically (Spinreact, Santa Coloma, Sant Esteve De Bas, Spain) [Kaplan, 1984], total cholesterol, triglycerides and high-density lipoprotein cholesterol (Pointe Scientific, Inc., Canton, Ohio, USA) [Fossati and Prencipe, 1982; Lopes-Virella et al. 1977; Allain et al. 1974]. Hemoglobin A1c concentrations were analyzed by the cation-exchange resin method (Hospitex Diagnostics, Sesto Fiorentino, Italy) [Trivelli, 1971]. Insulin was also analyzed by the Immulite 1000 kit (Siemen Medical Solution Diagnostics, Los Angeles, California, USA) [Sapin et al. 1998].
DNA genotyping
The E23K of Kir6.2 and G972R of IRS-1 polymorphisms were genotyped using the polymerase chain reaction-restricted fragment length polymorphism (PCR-RFLP) method. Genomic DNA was isolated from 200 µl whole blood samples using the Qiagen Extract kit (Qiagen, Hilden, Germany).
Kir6.2 genotyping
The DNA of the Kir6.2 gene was amplified by the PCR technique using a forward primer (5′-GAATACGTCCTGACACGCCT-3′) and a reverse primer (5′-GCCAGCTGCACAGGA AGGACAT-3′) (Operon, Ebersberg, Germany), which flanked the region containing the Kir6.2 gene (product size, 218 bp). PCR was performed using a 25 µl volume containing 5 µl genomic DNA, 1.5 µl of each primer, and 17 µl of the Taq PCR master mix kit (Qiagen). PCR conditions were denaturation at 95°C for 3 min, 35 cycles of denaturation at 95°C for 1 min., annealing at 60°C for 1 min, elongation at 72°C for 1 min, and a final elongation step at 72°C for 9 min.
Ten microliters of PCR product was digested with 2 µl of 10 U/µl BanII enzyme (Promega, Madison, Wisconsin, USA) at 37°C overnight. The digested probes were subjected to electrophoresis on 1% agarose (Sigma Chemical Co., St Louis, Missouri, USA) with 3% Nusieve agarose (Sigma Chemical Co.) and stained with ethidium bromide (Sigma Chemical Co.) for visualization. The gel was run at 150 V for 1 h [Ezenwaka et al. 2005].
IRS-1 genotyping
Two oligonucleotide primers: forward 5′-CTTCTGTCAGGTGTC CATCC-3′ and reverse 5′-TGGCGAGGTGTCCACGTAGC-3′ (Operon) were used for amplification of DNA which was carried out on 3 µl genomic DNA with a final volume of 25 µl. The assay consisted of 15 µl of Taq PCR master mix kit (Qiagen), and 2 µl of the two oligonucleotide primers. A cycling program using a GeneAmp 9600 thermal cycler (Progene, Tecnhe, Cambridge, UK) was as follows: initial denaturation at 95°C for 5 min, followed by 37 cycles of denaturation at 95°C for 45 s, annealing at 58°C for 45 s, elongation at 72°C for 45 s, and a final elongation step at 72°C for 5 min. Restriction enzyme digestion was carried out in 30 µl reactions containing 25 µl PCR product, 3 µl of 10× reaction buffer, 2 µl of the restriction enzyme MvaI. The reactions were incubated at 37°C overnight before electrophoresis. Electrophoresis was carried out on a 3% agarose gel and visualized after staining with ethidium bromide [Pavlova et al. 2004; Federici et al. 2003].
Statistical analysis
Continuous data were shown as means ± standard deviation (SD) and logarithmic transformation was used for data not normally distributed. Comparisons between genotypes were made using the unpaired Student’s t-test (E/E versus E/K + K/K and G/G versus G/R + R/R). Fisher’s exact test was used to test for differences in genotype frequencies with 95% confidence interval. The values at p < 0.05 were chosen as statistically significant. All analyses were performed using the Statistical Package of the Social Sciences (SPSS) software program.
Results
The clinical characteristics of the study participants stratified according to therapy and genotypes are reported in Table 1. Of all of the patients, 45% were carriers of the K allele. The genotype frequency of the K allele (E/K heterozygous + K/K homozygous) was 34% among patients with diabetes whose condition was controlled with sulfonylurea and 56% among patients with secondary failure to sulfonylurea (Figure 1). Thus, the E23K variant was associated with secondary failure to sulfonylurea with carriers of the K allele having a relative risk of 1.65 (95% CI 1.04–2.6, p = 0.04) compared with those who were E23E homozygous (Table 2).
The genotype frequencies of the E23K and Arg972 insulin receptor substrate-1 (IRS-1) variants among patients with secondary failure to sulfonylurea (group I) and patients with diabetes whose condition is controlled by sulfonylurea (group II). *p < 0.05: significant difference. Anthropometric and biochemical characteristics of the study population. Data are mean ± standard deviation. p < 0.05: significant difference. BMI, body mass index; HDL, high-density lipoprotein; HOMA-IR and HOMA-β cell, homeostasis model assessment of insulin resistance and of β-cell function respectively; LDL, low-density lipoprotein. QUICKI, quantitative insulin check index. Anthropometric, and biochemical characteristics of the study population according to the potassium inwardly rectifier 6.2 (Kir6.2) genotype. Data are means ± standard deviation. Differences between means were compared using unpaired Student’s t-test. p < 0.05: significantly different. BMI, body mass index; HDL, high-density lipoprotein; HOMA-IR and HOMA-β cell, homeostasis model assessment of insulin resistance and of β-cell function respectively; LDL, low-density lipoprotein. QUICKI, quantitative insulin check index.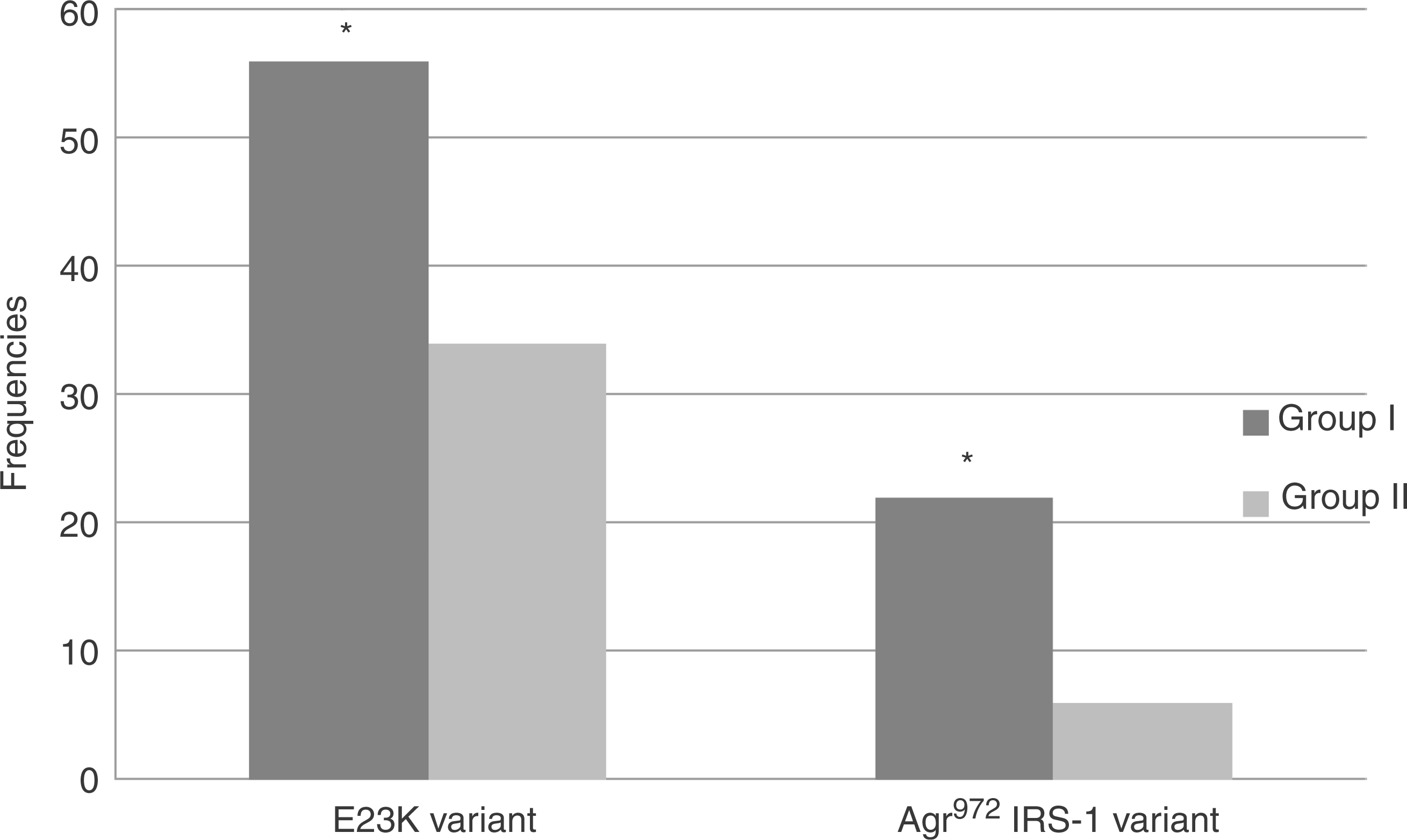

Anthropometric and biochemical characteristics of the study population according to the insulin receptor substrate-1 (IRS-1) genotype.

Data are mean ± standard deviation. Differences between means were compared using unpaired Student’s t-test.
p < 0.05: significantly different.
BMI, body mass index; HDL, high-density lipoprotein; HOMA-IR and HOMA-β cell, homeostasis model assessment of insulin resistance and of β-cell function respectively; LDL, low-density lipoprotein. QUICKI, quantitative insulin check index.
Discussion
This study investigated the relationship between the E23K variant and the risk for secondary failure to sulfonylurea in Egyptian patients with type 2 diabetes. The study found that the E23K variant was associated with secondary failure to sulfonylurea in patients carrying this variant compared with those who were noncarriers. This finding is supported by the study of Sesti and colleagues [Sesti et al. 2006] who reported a higher proportion of K-allele carriers (51.4%) among 208 patients whose condition failed to respond to sulfonylurea-metformin combination therapy and requiring insulin due to uncontrolled hyperglycemia (fasting plasma glucose >300 mg/dl). The data reported here are in contrast with the Prospective Diabetes Study (UKPDS) [Gloyn et al. 2001] in which the authors did not find any significant association between the E23K variant and response to sulfonylurea therapy. It is possible that differences in the definition of secondary sulfonylurea failure might account for these divergent results. In the UKPDS report, the authors identified patients whose condition failed to respond to sulfonylurea therapy when their fasting plasma glucose rose to more than 300 mg/dl. Also, the type of sulfonylurea (chlorpropamide in the UKPDS versus glibenclamide in the present study), and the clinical characteristics of patients (newly diagnosed patients aged 25–65 years in the UKPDS versus patients with known diabetes diagnosed at over 40 years of age in the present study).
The present study demonstrated that the E23K polymorphism is not associated with β-cell dysfunction as estimated by HOMA-β-cell function. Our observation is consistent with the other studies which provide preliminary evidence that the association of this polymorphism with increased blood glucose levels found in some other studies may be explained by diminished suppression of glucagon secretion in response to hyperglycemia [Tschritter et al. 2002]. This is at variance with results of the studies showing that the E23K polymorphism is associated with impaired glucose-induced insulin release [Hansen et al. 2005; Nielsen et al. 2003; Hart et al. 2002]. Villareal et al. (2009) reported that the Kir6.2 E23K variant is associated with multiple insulin secretory defects, including a reduction in overall β-cell responsiveness to glucose and a rightward shift in the dose–response curve between glucose and insulin secretion [Villareal et al. 2009].
Furthermore, the study found that the E23K variant was not associated with worsening metabolic control (fasting glucose level, fasting serum insulin, and hemoglobin A1c) across genotyping groups. Our observation is consistent with the studies which found that the E23K polymorphism does not increase impaired glucose tolerance in carriers of the E23K polymorphism [Ezenwaka et al. 2005]. Jose and colleagues [Jose et al. 2007] observed no significant longitudinal changes in fasting glucose levels across genotypic groups in the metformin arm. This is in apparent contrast with the study of Sesti and colleagues who found that the E23K variant was associated with worsening metabolic control in the subgroup of patients whose condition was controlled with sulfonylurea [Sesti et al. 2006]. Holstein and colleagues suggested that patients with type 2 diabetes carrying the K allele have reduced response to sulfonylurea therapy, which results in increased hemoglobin A1c [Holstein et al. 2009].
It should be noted that the number of patients in the present study is relatively small and this might have reduced the statistical power of the tests. However, the results have demonstrated that the presence of the Kir6.2 E23K genotype in Egyptian patients with diabetes did not increase fasting serum glucose levels or glycoslated hemoglobin percentage. This study and others have found that the estimated insulin resistance by HOMA and insulin sensitivity by QUICKI showed a weak association of the K allele with impaired insulin resistance and sensitivity [Jose et al. 2007; Tschritter et al. 2002]. This is at variance with the results of a study demonstrating an association of the homozygous K allele with impaired insulin sensitivity [Wan et al. 2009]. Analysis of the present data did not show any significant differences in the biochemical risk factors for developing diabetes in patients who were obese and those who were non-obese with the E23K variants, which is consistent with previous reports [Jose et al. 2007; Ezenwaka et al. 2005]. However, many studies found that the E23K variant is associated with a significant increase in BMI, suggesting that this polymorphism may be diabetogenic through obesity-related mechanisms [Nielsen et al. 2003].
The study also investigated the association between a common polymorphism in IRS-1 causing a Gly972Arg change and secondary failure to sulfonylurea and found that the Arg972 IRS-1 variant was associated with secondary failure to sulfonylurea. Sesti and colleagues supported these results with the notion that the Arg972 IRS-1 variant impairs insulin secretion induced by secretagogues other than glucose [Sesti et al. 2004]. Chronic activation of insulin receptor signaling by IRS-1 overexpression in the β-cell inhibited gene expression of endoplasmic reticulum Ca(2+)-ATPase and subsequent inhibition of Ca+2 uptake leading to an increase in cytosolic Ca+2 concentrations [Xu et al. 2000]. As reported above, and consistent with the results of Stumvoll and colleagues, patients with diabetes carrying the Arg972 IRS-1 polymorphism have a lower β-cell function level, expressed in HOMA-β, than that of patients carrying the wild-type IRS-1 variant in both groups (p = 0.02, 0.045 respectively) [Stumvoll et al. 2001]. The markedly reduced insulin secretion was accompanied by a decreased binding of the p85 subunit of PI3-K to IRS-1. Marchetti and colleagues showed that human pancreatic islets from carriers of the G972R amino acid polymorphism in IRS-1 have reduced insulin content, altered insulin release, and greater number of immature secretory granules [Marchetti et al. 2002]. In vitro studies have suggested that pancreatic islets isolated from carriers of Arg972 IRS-1 exhibit impaired IRS-1-associated PI 3-kinase activity and increased apoptosis. They also appear resistant to the antiapoptotic effect of insulin compared with wild-type controls and so contribute to the risk for secondary failure [Federici et al. 2001]. In contrast, Laakso and colleagues did not find any significant association of the Gly–Arg972 substitution with maximum insulin secretion capacity in an oral glucose tolerance test in patients with type 2 diabetes [Laakso et al. 1994].
This study has demonstrated that the Arg972 variant contributes to increase in fasting serum insulin levels among patients with diabetes well controlled with oral therapy and among patients with secondary failure to sulfonylurea (p = 0.009 and 0.01, respectively). Subsequently a significant increase in insulin resistance, represented by HOMA-IR (p = 0.03 and 0.002, respectively), and a decrease in insulin sensitivity, represented by QUICKI (p = 0.03 and 0.003, respectively), were observed.
The present results are in agreement with several other studies [Lin et al. 2006; Villuendas et al. 2005]. McGettrick and colleagues investigated the detailed molecular mechanism by which this polymorphism may be linked to insulin resistance. They demonstrated that the Arg972 variant alters the ability of the recombinant IRS-1 (925–1008) fragment to be phosphorylated at specific tyrosine residues flanking the polymorphism, which normally form high-affinity binding sites for the p85α regulatory subunit of PI 3 kinase [McGettrick et al. 2005]. Other investigations have failed to observe the association between the Arg972 IRS-1 polymorphism and insulin resistance [Koch et al. 1999; Laakso et al. 1994]. Differences in ethnic background and the technique used to assess insulin resistance may explain this discrepancy.
Moreover, the comparably increased prevalence of the Arg972 variant in patients with obesity in groups I and II (p = 0.009 and 0.03, respectively) is consistent with a role of the Arg972 variant in causing insulin resistance in these patients. The same results were reproduced in the study of Baroni and colleagues [Baroni et al. 2001]. Other authors have not found an association between obesity and the Arg972 variant [Srivastava et al. 2008].
This study found that the most remarkable change in the lipid profile of mutation carriers in both groups was the increase in plasma total cholesterol (p = 0.02, 0.04, respectively) and low-density lipoprotein cholesterol (p = 0.007, 0.03, respectively). Abe and colleagues described significantly higher triglyceride levels in heterozygous carriers of the Arg972 variant [Abe et al. 1998]. In this respect, IRS-1-mediated activation of PI 3 kinase has been reported to be involved in the antilipolytic effect of insulin. Because it has been demonstrated that the G972R substitution significantly reduces the IRS-1-mediated PI 3-kinase activation, it is conceivable that mutation carriers may have impaired antilipolysis. As a consequence, an increased efflux of free fatty acid from adipose tissue would provide more substrate available for very-low-density lipoprotein triglyceride synthesis by the liver [Baroni et al. 1999].
Overall, these results confirm the growing body of evidence reflecting the association between both the E23K variant in the Kir6.2 gene and the Arg972 variant in the IRS-1 gene and increased risk for secondary failure to sulfonylurea in patients with type 2 diabetes. The documented and reproducible association of selected polymorphisms in genes that encode drug targets with type 2 diabetes highlights the possible application of human genomics to medicine.
Footnotes
Acknowledgements
All of the staff at the Clinical Immunology Department, Mansoura University, Medicine College are gratefully acknowledged for fruitful co-operation.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The author declares no conflict of interest in preparing this article.