
Abstract
Fibroblast growth factor (FGF) signaling plays essential roles in various biological events. FGF18 is one of the ligands to be associated with osteogenesis, chondrogenesis and bone healing. The mouse critical-sized calvarial defect healing induced by the bone morphogenetic protein 2 (BMP2)-hydrogel is stabilized when FGF18 is added. Here, we aimed to investigate the role of FGF18 in the calvarial bone healing model. We first found that FGF18 + BMP2 hydrogel application to the calvarial bone defect increased the expression of anti-inflammatory markers, including those related to tissue healing M2 macrophage (M2-Mø) prior to mineralized bone formation. The depletion of macrophages with clodronate liposome hindered the FGF18 effect. We then examined how FGF18 induces M2-Mø polarization by using mouse primary bone marrow (BM) cells composed of macrophage precursors and BM stromal cells (BMSCs). In vitro studies demonstrated that FGF18 indirectly induces M2-Mø polarization by affecting BMSCs. Whole transcriptome analysis and neutralizing antibody treatment of BMSC cultured with FGF18 revealed that chemoattractant chemokine (c-c motif) ligand 2 (CCL2) is the major mediator for M2-Mø polarization. Finally, FGF18-augmented activity toward favorable bone healing with BMP2 was diminished in the calvarial defect in Ccr2-deleted mice. Altogether, we suggest a novel role of FGF18 in M2-Mø modulation via stimulation of CCL2 production in calvarial bone healing.
Introduction
Calvarial defects caused by congenital anomalies, traumatic injuries, or tumors often require surgical treatment such as bone grafting, to optimize bone repair for critical-sized defects. 1 Bone healing involves multi-step processes and crosstalk between osteogenesis-associated cells and immune cells.2,3 According to the current concept in tissue engineering, the combination of growth factors, cells, and biocompatible materials/scaffolds has been the key to exploring and initiating suitable strategies for favorable outcomes. 4 We have focused on utilizing growth factors in conjunction with hydrogel, a biocompatible and biodegradable material, as a delivery system in bone engineering.
Numerous signaling pathways including Wnt, Notch, fibroblast growth factor (FGF), and bone morphogenetic protein (BMP) signalings, function in coordination during bone development, remodeling, and repair processes. 2 Among these pathways, bone morphogenetic protein 2 (BMP2) is considered the gold standard for bone formation and healing, officially approved as an osteoinductive growth factor to substitute bone grafts. However, the high dosage of BMP2 required for optimal healing can lead to unwanted side effects such as increased post-operative inflammation. 5 The incorporation of additional growth factors might emerge as a promising strategy to mitigate the adverse effects associated with high BMP2 dosage. This approach holds the potential to improve the overall therapeutic outcome and establish the safer strategy for bone healing treatment.
Of particular interest, FGF signaling can be a promising pathway for that approach. FGF ligands play fundamental roles in controlling cell growth, cell differentiation, and mediating tissue repair and regeneration by activating intracellular cascades through their receptors, FGFR 1-4. 6 The significance of FGF signaling in bone biology is highlighted by the skeletal abnormalities observed in various FGF/FGFR mutations, including craniosynostosis, the premature fusion of cranial sutures.6,7 One member of the paracrine FGF ligands, FGF18, belongs to the FGF8 subfamily. 6 FGF18 has been identified as a positive regulator of osteogenesis, which is demonstrated by the smaller cranial vault and retarded cranial bone growth observed in Fgf18-deleted mice.8,9 When FGF18-soaked beads were applied to developing mouse fetal calvaria, osteogenesis was accelerated along with an upregulation of Bmp2 expression. 10 Furthermore, it has been shown that FGF18 exhibits an osteoinductive activity on mesenchymal stromal cells (MSC) and contributes to skull bone formation.11–13 Thus, FGF18 could be a potential candidate for supporting and promoting BMP2-dependent bone healing.
We previously reported that FGF18 enhances the osteogenic properties of BMP2 by improving the healing rate of critical-sized mouse parietal bone defects.14,15 When FGF18 was added to the amount of BMP2 that resulted in various degrees of healing with 71.0% defect closure on average, it significantly improved the healing rate to 93.4% on average after 8 weeks. Moreover, the addition of FGF18 led to the formation of good quality and quantity of new bones with bone marrow-like structure, 14 which suggested the remodeling potential. Of note, a single application of FGF18 alone was not sufficient to stimulate favorable bone formation, unlike the application to developing bones in fetal period. Therefore, further investigation is needed to understand the mechanism behind the stabilizing effect of FGF18 on BMP2-dependent bone healing.
Interestingly, recent findings have shed light on the involvement of the FGF signaling pathway in the regulation of inflammation and macrophage activation. Macrophages are effective immune cells that participate in various phases of inflammation to eradicate pathogens and promote tissue healing.16–18 Additionally, macrophages are essential in tissue repair, tissue regeneration, maintaining homeostasis, and immune surveillance in numerous tissues including bone.19,20 Macrophages are mainly classified into two subpopulations based on the function, M1 and M2 macrophages, distinguished by cell surface markers and biological activities. M1 macrophages (M1-Mø) display a pro-inflammatory phenotype,21,22 whereas M2 macrophages (M2-Mø) exhibit pro-healing, anti-inflammatory, and reparative properties. M2-Mø also play a pivotal role to create a favorable bone healing outcome. 23 FGF1-2 and FGFR1, 3 exert both pro-inflammatory and anti-inflammatory functions depending on the specific tissue context and body response. 24 FGF2 induces M2-Mø polarization of tumor-associated macrophages, in vivo. 25 FGF9 was reported to stimulate M2-Mø activation and enhance the production of anti-inflammatory cytokine in infarcted heart of diabetic mice. 26 FGF21 demonstrated the ability to reduce M1-Mø activation in mouse model of ischemic stroke. 27 Moreover, FGF18 has emerged as a promising therapeutic target for treating osteoarthritis due to its involvement in maintaining chondrocyte viability and reducing the synthesis of pro-inflammatory factors. 28 Thus, FGF signaling has the potential to control the inflammatory process and it is expected that FGF18 could be involved in the immunomodulation of macrophages during process of stabilization of BMP2-dependent bone healing.
However, the immunomodulatory effect of FGF18 on macrophages during calvarial defect healing has not yet been clarified. We hypothesized that FGF18 creates the anti-inflammatory microenvironment that enhances bone healing by modulating macrophage activity to M2-Mø phenotype. In this study, we investigated the effect of FGF18 on inflammatory status during the early stage of bone healing using an in vivo mouse calvarial bone defect model and carried out a series of in vitro experiments to uncover the novel function of FGF18 with bone marrow cells. Our findings provide important knowledge about how FGF18 modulates macrophages and suggest the potential of FGF18 as an adjunctive growth factor in bone regeneration, leading to significant therapeutic outcomes.
Materials and methods
Animal study
All animal experiments were approved by the Institutional Animal Care and Use Committee of Tokyo Medical and Dental University (A2018-48C, A2019-060A, A2021-198A, A2021781). C57BL/6J wild-type mice (Sankyo Labo Service Corporation, Japan) and chemokine (c-c motif) receptor 2 homozygous knockout (Ccr2 -/- ) mice (The Jackson Laboratory, USA) were maintained in specific pathogen-free (SPF) condition with ad libitum food and water according to the regulations and guidelines of Center for Experimental Animals, Tokyo Medical and Dental University.
Preparation of CHPOA NanoClik hydrogel
Cholesteryl group- and acryloyl group-bearing pullulan (CHPOA), which contains 23.1 acryloyl groups per 100 anhydrous glucoside units, was synthesized in-house. The CHPOA Nano-Crosslinked (NanoClik) hydrogel was prepared following a previous report. 14 In brief, the CHPOA nanogel was re-suspended in sterile phosphate buffer saline (PBS) at a concentration of 33 mg/ml overnight. It was then mixed with recombinant human fibroblast growth factor 18 (FGF18, Peprotech Inc., USA, Cat.100-28) and/or recombinant human bone morphogenetic protein 2 (BMP2, R&D Systems Inc., USA, Cat.355-BM-100/CF), or with PBS only, and incubated at 4°C for 24 h to generate the CHPOA-growth factor nanogel complex. Next, the CHPOA nanogel complex was mixed with pentaerythritol tetra (mercaptoethyl) polyoxyethylene (PEGSH/PTE-100SH, NOF Corporation, Japan, Cat.188492-68-4) at a concentration of 640.8 mg/ml. A mixture of 4.2 µl was then dropped onto a Parafilm® M-coated glass slide (Matsunami Glass Ind. Ltd., Japan) to fabricate a circular-shaped hydrogel disk with a thickness of 0.5–0.6 mm and a diameter of 3 mm. The hydrogel disk was maintained at 37°C with a humidified atmosphere for 24 h. Thus, a CHPOA/PEGSH NanoClik hydrogel disk contained 500 ng of FGF18 and BMP2 each (FGF18 + BMP2), or 500 ng of BMP2 (BMP2).
Calvarial defect surgery
Four-week-old wild-type or Ccr2 -/- mice (16–20 g of body weight) were anesthetized by intraperitoneal administration of 3-mixed anesthetic agents 29 of medetomidine (Domitor, Orion Corporation, Finland), midazolam (Dormicum, Maruishi Pharmaceutical Co., Ltd., Japan) and butorphanol (Vetorphale, Meiji Seika Pharma Co., Ltd., Japan) in PBS at 5 µl/g body weight. The calvarial defect surgical operation was performed as previously reported. 15 A vertical full-thickness skin flap incision was made along the mid-sagittal line of the shaved scalp. The periosteum overlying the cranium was gently retracted to the lateral sides. A critical-sized circular cut (3 mm in diameter) 30 was gently made on the parietal bone by using a disposable biopsy punch (Kai industries Co., Ltd., Japan). The bone piece was carefully removed not to damage the dura mater, and bleeding was stopped with sterile gauze pellets. The defect was then applied with a CHPOA NanoClik hydrogel disk, covered with periosteum, and the skin flap was sutured with 4.0 silk using 3/8 needle (Matsuda, Japan). After surgery, operated mice received intraperitoneal injection of atipamezole (Antisedan, Orion Corporation, Finland) in PBS (dose 5 µl/g body weight) 29 and housed in warm environment for recovery. Operated mice were maintained for 5–7 days before the tissue collection from the defect area and histological analysis (n = 4 per group), or for 4–8 weeks post-surgery for radiological assessment of calvarial bone healing (n = 3–5 per group).
Calvarial defect collection
Operated mice with PBS, BMP2, or FGF18 + BMP2 NanoClik hydrogel implantation were sacrificed at day 5 post-surgery. The calvarial bone was dissected in RNase-free iced-cold PBS (n = 4 per group) by removing the overlying skin, periosteum, and brain. The tissue sample was harvested by trimming the bone 0.5–1 mm from the original defect border using micro-scissors under a stereomicroscope (Olympus SZX7, Japan). This defect sample includes the bone defect rim with some hydrogel and the dura mater. The samples were homogenized in Sepasol-RNA I Super G (Nacalai Tesque Inc., Japan) using a disposable homogenizer (BioMasherII, Nippi, Japan), and stored at −80°C before RNA extraction.
For histological analysis, operated mice were sacrificed at 1 week after surgery. The calvaria were rapidly dissected by peeling off the skin. After trimming, the samples were fixed in 4% paraformaldehyde (PFA) (Nacalai Tesque Inc., Japan) in PBS (4% PFA/PBS) containing 20% sucrose for 3 h, then embedded in SCEM-L1 (Section-Lab, Japan) and frozen at −80°C (n = 3 per group).
Histological analysis
For undecalcified calvarial sections, frozen samples were cryosectioned at 6 μm thickness (Leica RM2235, Germany) by the Kawamoto’s film method 31 (n = 3 per group). Sections were stained by Mayer’s Hematoxylin and 1% Eosin Y solution. For alkaline phosphatase and alizarin red staining, the sections were washed twice with alkaline phosphatase buffer (0.1 M Tris-HCl pH 9.5 + 0.1 M NaCl + 0.05 M MgCl2) at room temperature for 3 min. Then, the sections were incubated in nitro blue tetrazolium chloride (NBT)/5-bromo-4-chloro-3-indolyl-phosphate (BCIP) solution (Roche, USA) for 5 min, followed by fixation in 4% PFA/PBS for 5 min. The sections were counterstained with 0.5% Alizarin Red S solution (SIGMA, USA, A5533) for 5 min. Sections were visualized using a BZ-X700 microscope (KEYENCE, USA)
For immunofluorescent staining, antigen was retrieved in HistoVT One (pH 7.0) (Nacalai Tesque Inc., Japan) in Milli-Q water at 90°C for 20 min. After non-specific blocking with 5% horse serum (Vector Laboratories, USA, S-2000) in 0.1% Tween (Nacalai Tesque Inc., Japan) in PBS, phycoerythrin (PE)-conjugated anti-mouse F4/80 monoclonal (dilution 1:200, eBioscience, USA, Ref 12-4801-82) and Alexa Fluor® 488-conjugated anti-mouse CD206 (1:250, BioLegend, USA, Cat.141710) antibodies were applied on sections. Nuclear counterstaining was performed using Hoechst 33342 solution in PBS (1:1000, Dojindo, Japan, Cat.23491-52-3) (n = 3). Immunofluorescent signals were detected and localized using a BZ-X700 microscope (KEYENCE, USA) or a Leica SP8 confocal microscope (Germany). The number of CD206+ M2-Mø were counted from six different areas around the calvarial defect obtained from two different sagittal section planes (central and lateral regions) using ImageJ software (National Institutes of Health, USA).
RNA preparation and reverse transcription polymerase chain reaction (RT-PCR)
Calvarial tissues or cells were homogenized using Sepasol-RNA I Super G and total RNA was extracted according to the product protocol. Total RNA (1 µg) was reverse transcribed to complimentary DNA (cDNA) by ReverTra Ace® (Toyobo Co. Ltd., Japan) following manufacturer’s instruction.
Semi-quantitative RT-PCR was performed with T100™ Thermal cycler (Bio-Rad Laboratory Inc., USA). DNA fragments of the gene of interest was amplified by GoTaq® Green Master Mix (Promega Corporation, USA) following the manufacturer’s protocol. PCR products were detected by electrophoresis in 3% Tris-Acetate-EDTA agarose gel (Nippon Gene, Japan) containing 1% v/v ethidium bromide (Nippon Gene, Japan) and the bands were visualized under ultraviolet light. Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) was used as the internal control gene. cDNA samples from independent batches of activated tissues or cells were used (n = 3).
Quantitative RT-PCR (RT-qPCR) was performed with KOD SYBR® qPCR Mix (Toyobo Co.Ltd., Japan) and assessed by LightCycler® 96 System (Roche Diagnostics, USA) according to manufacturer’s guideline. The mRNA expression levels were presented as relative mRNA expression normalized by that of Gapdh and analyzed by ΔΔCt method. The experiments were conducted in triplicate and repeated at least four times using cDNA from independent batches of activated tissues or cells (n = 4–5). Oligonucleotide sequences for target genes in RT-qPCR (Thermo Fisher Scientific Inc., USA) are shown in Supplemental Table S2.
In vivo macrophage depletion
Clodronate liposome (Clophosome N, FormuMax Scientific Inc., USA, Cat.F70101C-N-10) was intravenously (I.V.) injected into the operated mice at dose of 8 µl/g body weight via the lateral tail vein immediately after the surgical procedure for 1-week Mø-depletion. Some of the mice received another injection at day 7 for 2-week Mø-depletion. The control group received an injection of PBS liposome (FormuMax Scientific Inc., USA) (n = 5 per group).
Micro-computed tomography (µCT) analysis
Radiological imaging was performed using Micro Focus X-Ray CT System (inspeXio SMX100CT, Shimadzu Corporation., Japan). Each mouse was anesthetized and subjected to μCT scanning at 0, 1, 2, 3, 4, and 8 weeks post-surgery (n = 3–5 per group). Reconstruction for three-dimensional visualization was obtained from the volume-rendering software (VGStudio MAX 2.0, Volume Graphics, Germany) and the two-dimensional image of defect was exported for analysis. Standardized images of calvaria were evaluated and the defect area was measured with Photoshop CC 2019 (Adobe, USA). The bone healing was calculated as the percentage of the newly formed bone area to the initial defect area on the operation day (0 week). 14
Isolation of bone marrow cells and induction of bone marrow-derived macrophage differentiation
Bone marrow (BM) cells were obtained from the tibial and femoral medullary cavity of 8–12 weeks old wild-type mice after euthanasia procedure following the previous method 32 with minor modifications. In brief, the tibia and femur bones were dissected, and the skin and muscles were removed. The bones were then cut at the ankle joint, the knee joint, and the pelvic-to-hip joint in iced-cold PBS supplemented with 10 µg/ml gentamicin sulfate (Nacalai Tesque Inc., Japan), and bone samples were kept in ice-cold culture medium. BM was flushed from medullary cavity using a 25 -G needle (Terumo Corporation, Japan), erythrocyte lysis was carried out with 0.83% ammonium chloride, and centrifuged at 300×g at 4°C for 10 min to obtain total BM cells.
Total BM cells were plated on the tissue culture dish (Corning Inc., USA) in complete DMEM medium, which is Dulbecco’s Modified Eagle Medium (DMEM) (high glucose + 110 µg/ml sodium pyruvate) medium (Fujifilm Wako Pure Chemical Corporation; Wako, Japan) containing 10% fetal bovine serum (FBS) (Gibco, Life Technologies Corporation, USA), 2 mM L-glutamine (Wako, Japan) and 10 µg/ml gentamicin sulfate. After overnight culture, non-adherent cells were collected as BM progenitor cells for further assays. The in vitro protocol is illustrated in Supplemental Figure S1.
BM progenitor cells (4 × 106 cells) were seeded on the petri dish (Corning Inc., USA) in macrophage complete medium, which is complete DMEM medium supplemented with 20 ng/ml recombinant human macrophage colony stimulating factors (M-CSF, Wako, Japan). The cells were incubated in a humidified incubator with 5% CO2 at 37°C for 7 days and culture medium was refreshed every 3 days. Only BM progenitor cells attached and differentiated into macrophages on the surface of petri dish.
On day 7, non-polarized BM-derived macrophages (M0-Mø) were harvested by non-enzymatic cell dissociation solution (Biological Industries, Israel) and characterized by the expression of F4/80, CD11b and CD206 proteins using flow cytometry.
Macrophage activation
BM progenitor cells were cultured on the petri dish and maintained in complete macrophage medium supplemented with 1 or 10 ng/ml FGF18 for 3 or 7 days. In other groups, M0-Mø or RAW264.7 cells (mouse macrophage cell line) were seeded on the 24-well plate at density 4×105 cells/well and incubated overnight. These macrophage cultures were then maintained in complete DMEM medium supplemented with 1 or 10 ng/ml FGF18 for 24–72 h. Treatment with 20 ng/ml recombinant murine interleukin-4 (IL-4, Peprotech Inc., USA, Cat.214-14) was used for in vitro M2-Mø polarization in all culture systems. This experiment was carried out by using primary cells from independent mice (n = 3–4).
Total bone marrow cell culture and activation
The isolated total BM cells were cultured in the 24 well-plate at density 3.75 × 105 cells/well (Corning Inc., USA) with or without a 15 mm circular micro-cover glass (Matsunami Glass Ind. Ltd., Japan) in the macrophage complete medium. FGF18 at 1 or 10 ng/ml or IL-4 at 20 ng/ml was added either from day 0 (beginning point) or on day 4 of 7-day culture period. This culture system allows M0-Mø and other non-hematopoietic BM-resident cells such as BM stromal cells, to attach and proliferate or differentiate on the surface. On day 7, the activated cells were subjected to immunofluorescence or RT-PCR analyses. This experiment was carried out using three independent total primary BM cells (n = 3).
Flow cytometry
Activated M0-Mø were collected and prepared for flow cytometry analysis. Cells were washed twice with PBS containing 0.2% azide, treated with culture supernatant from 2.4G2 (anti-CD16/CD32 mAb) for non-specific blocking of FcγR and stained with macrophage-specific fluorochrome-conjugated monoclonal antibodies, including PE-conjugated anti-mouse F4/80 monoclonal antibody (1:8), allophycocyanin (APC)-conjugated anti-mouse CD11b antibody (1:40, eBioscience, USA, Ref 17-0112-8), and Alexa Fluor® 488-conjugated anti-mouse CD206 antibody (1:20). Stained cells were analyzed using a FACS caliber flow cytometer (Bio-Rad Laboratory Inc., USA). The percentage of F4/80+CD11b+CD206+ M2-Mø in FGF18- or IL-4-treated groups was normalized to that of control group and represented as fold change of M2-Mø percentage. The samples collected from three independent animals (n = 3).
Immunofluorescence of BM cells
Cells were fixed with 4% PFA/PBS for 10 min and then permeabilized with 0.1% TritonX-100 (Nacalai Tesque Inc., Japan) in PBS for 5 min at room temperature. Non-specific blocking was carried out by 2% bovine serum albumin in PBS (Sigma-Aldrich, USA) for 1 h. Next, fluorophore-conjugated monoclonal antibodies were added, including PE-conjugated anti-mouse F4/80 monoclonal antibody (1:200) and Alexa Fluor® 488-conjugated anti-mouse CD206 antibody (1:500), and incubated at 4°C overnight. Nuclear counterstaining was performed with Hoechst 33342 solution in PBS (1:1000)
Stained cells were visualized by a DM5000B fluorescent microscope (Leica, Germany) and images were captured by Leica DFC310Fx (Germany). The number of F4/80+ cells and F4/80+CD206+ cells were manually counted in at least three different randomly selected images using ImageJ software. The percentage of F4/80+ macrophages and CD206+ M2-Mø were calculated by the ratios of total F4/80+ cells to all nuclei and total F4/80+CD206+ cells to total F4/80+ cells, respectively. This experiment was implemented by using three independent sets of BM cells from different animals (n = 3).
Cell proliferation assay
M0-Mø or RAW264.7 cells (5 × 103 cells/well) were seeded on the 96-well plate and incubated with complete DMEM medium overnight. The complete DMEM medium supplemented with FGF18 at 1 or 10 ng/ml was added to each well at 0 h. At 24, 48, 72 or 96 h of culture, cell counting kit-8 (CCK-8, Dojindo, Japan) solution was added and incubated for 2 h at 37°C following manufacturer’s instruction. Then, incubated CCK-8 solution was collected, and the absorbance was measured at 450 nm using an ARVO™ MX 1420 multilabel counter (PerkinElmer, USA). This experiment was performed in triplicate and repeated at least three times (n = 3–4).
Cell migration assay
RAW264.7 cells were seeded on the 6-well plate at density 1 × 106 cells/well and maintained with complete DMEM medium until 100% confluence. Cells were starved in FBS-free medium for 4 h, then horizontal scratch was made by a 200 µl pipette tip, washed detached cells with PBS and replaced with FBS-free DMEM medium supplemented with 1 or 10 ng/ml FGF18. Migrating cells into scratched wound area were observed and the images were captured at 0, 8, and 24 h with an inverted microscope (Olympus CKX53 with DP20, Japan). The closing wound area was measured by ImageJ software (n = 5).
Bone marrow stromal cell-conditioned medium (BM-CM) and activation
Primary BM stromal cells (BMSCs) were cultured with Minimum Essential Medium alpha (MEM-α) (Wako, Japan) supplemented with 10% FBS and 10 µg/ml gentamicin sulfate (complete MEM-α medium) as previous described 33 with some modifications. Non-adherent cells were periodically removed every 8 h during the first 3 days. Adherent BMSCs were washed with PBS on day 5, cultured until they reached to 80%–90% confluency at 37°C with 5% CO2 and passaged. Culture medium was refreshed every 3 days and cells obtained from passages 2 to 4 were used in further assays.
To generate conditioned medium (BM-CM), BMSCs were cultured in MEM-α with 0.1% FBS and 10 ng/ml FGF18. After 72 h, culture medium was collected and centrifuged at 2500×g for 5 min to remove cells and cell debris (oBM-CM). The oBM-CM was then filtered by an Amicon® Ultra-15 10 K centrifugal filter device (Merck Millipore Ltd., Ireland) following manufacturer’s instruction. Residual BM-CM (rBM-CM) was prepared by adding the same volume of MEM-α as the device-loaded volume of oBM-CM. All BM-CMs were kept in −80°C freezer. For macrophage activation, M0-Mø (cell density 4 × 105 cells/well) were seeded on the 24-well plate and incubated overnight. Subsequently, each type of BM-CMs supplemented with 10% FBS were added at 750 µl/well, and M0-Mø were cultured in a 37°C humidified incubator with 5% CO2 for 72 h (n = 4–5).
Microarray analysis
The BMSCs were cultured in MEM-α and 0.1% FBS with or without 10 ng/ml FGF18. After 24 h, cells were harvested and homogenized in SepaSol-RNA I Super G, and total RNA was further isolated. Microarray analysis was carried out using Clariom™ S Assay, mouse (Thermo Fisher Scientific Inc., USA) by Filgen Inc. (Japan) according to the manufacturer’s protocol (n = 6). Data were analyzed by the Expression Console™ Software (Thermo Fisher Scientific Inc., USA) and the target gene list was sorted by the Microarray Dara Analysis Tool (Filgen Inc., Japan) and Microsoft Excel (Microsoft Corporation, USA). RNA samples isolated from three independent mice were submitted for analysis (n = 3 per group).
M2 macrophage polarization by CCL2
M0-Mø (cell density 1 × 106 cells/well) were seeded on the 12-well plate and incubated overnight. Recombinant murine chemokine ligand 2 (CCL2, Peprotech Inc., USA, Cat.250-10) at 50 ng/ml were added to complete DMEM medium and maintained at 37°C with 5% CO2 for 24 h. M0-Mø culture maintained with rBM-CM was used for the control (n = 4–5).
CCL2 neutralization assay
Anti-mouse CCL2/MCP-1 rat IgG (R&D Systems Inc., USA, MAB479-SP) or rat isotype-matched control IgG (GeneTex Inc., USA, GTX35047) at final concentration 1 or 2 µg/ml was added to rBM-CM supplemented with 10% FBS, and incubated for 1 h at 37°C. M0-Mø were seeded on the 24-well plate in complete DMEM medium overnight and cultured with 750 µl/well of IgG isotype-, anti-CCL2 IgG-pretreated rBM-CM, FGF18-untreated rBM-CM or complete DMEM medium at 37°C with 5% CO2 incubator for 72 h (n = 4).
In vitro osteoclastogenesis
In vitro osteoclast differentiation was performed according to the previous methods.34,35 In monoculture system, bone marrow-derived monocyte/macrophage precursor cells were cultured in complete MEM-α supplemented with 10 ng/ml M-CSF (R&D system Inc., USA) for 2 days to obtain osteoclast precursors. The osteoclast precursor cells were further cultured with 10 ng/ml M-CSF and 25 ng/ml RANKL (Peprotech Inc., USA) in the presence of 2 or 20 ng/ml FGF18 for 3 days (n = 3). In co-culture system, BM cells were cultured with calvarial osteoblasts supplemented with 10 nM 1,25-dihydroxyvitamin D2 (1,25(OH)2D3, Wako, Japan) and 1 mM prostaglandin E2 (PGE2, Cayman Chemical, USA) for 7 days. FGF18 at 2 or 20 ng/ml was added in culture medium from day 0 (n = 3). Osteoclastogenesis was assessed by tartrate-resistant acid phosphatase (TRAP) staining. TRAP-positive multinucleated (more than three nuclei) cells were counted.
Statistical analysis
All experiments were conducted at least three times as indicated. All data were represented as mean and standard deviation (S.D.) or the boxplots and interquartile range. Statistical analysis was assessed using Prism9 (GraphPad software, USA). For two group comparison, the student’s t-test (two-tailed) were employed. For multiple comparison, one-way ANOVA with Tukey’s post-hoc test was performed. Statistical significance was defined at p value <0.05.
Results
FGF18+BMP2 hydrogel application promotes M2 macrophage polarization in calvarial bone defects
To examine the augmented function of FGF18 in BMP2-dependent bone healing in a critical-sized parietal bone defect model 14 (Figure 1(a)), we applied cholesteryl group- and acryloyl group-bearing pullulan (CHPOA) Nano-Crosslinked (NanoClik) hydrogel to 4-week-old mice (Figure 1(b)). Initially, we investigated the profile of inflammatory-related genes in the bony defect using real-time RT-PCR (RT-qPCR) at the early stage of healing. Since the newly formed mineralized bone first appears by 1 week post-surgery with FGF18 + BMP2 application, 14 we collected the thin bony rim of the defect with the dura mater from the operated mice at day 5 post-surgery (Figure 1(c)).

FGF18-augmented BMP2 activity on mouse calvarial defect healing involves macrophage modulation.
We found mRNA expression levels of pro-inflammatory cytokines, tumor necrosis factor alpha (Tnfa), and interleukin 1-beta (Il1b), as well as the M1-Mø marker gene, 22 nitric oxide synthase 2 (Nos2 or iNos), were significantly reduced in both the BMP2 and the FGF18 + BMP2 groups compared to the PBS group (Figure 1(d)–(f)). In contrast, several M2-Mø associated genes,21,22 including mannose receptor C-type 1 (Mrc1 or Cd206), arginase 1 (Arg1), chitinase-like 3 (Chil3 or Ym1), peroxisome proliferator-activated receptor gamma (Pparg), and the anti-inflammatory cytokine interleukin-10 (Il10), were elevated only in the FGF18+BMP2 group (Figure 1(g)–(k)). The osteogenesis related genes, Sp7 transcription factor (Sp7 or osterix) and alkaline phosphatase (Alpl) were also upregulated only in the FGF18+BMP2 group (Figure 1(l) and (m)). These results suggested that the addition of FGF18 enhanced M2-Mø polarization, which results in promotion of BMP2-induced calvarial bone healing.
Histological analyses on day 7 post-operation showed the significant increase in infiltration of CD206+ M2-Mø in the healing area of the FGF18 + BMP2 group compared to the other groups (Figure 1(n) and (o), Supplemental Figure S2A), Accordingly, the alkaline phosphatase (ALP)-positive osteoblasts were accumulated in the defect area including the meningeal layer (dura mater) in the FGF18 + BMP2 group (Supplemental Figure S2B). These observations strongly supported our hypothesis that FGF18 stimulates M2-Mø polarization to participate in the anti-inflammatory process and promote new bone formation.
Depletion of macrophages during early healing period demolishes FGF18-augmented effect on BMP2 osteogenic activity in calvarial defect
To confirm the involvement of FGF18 in macrophage modulation, we depleted the macrophage population during the early healing phase using clodronate liposome (Clo). The dose and timing of Clo injection were optimized based on the depletion efficacy and the health of operated mice. By treating mice with Clo at a dosage of 8 µl/g body weight, the number of F4/80+CD11b+ macrophages was notably reduced in the bone marrow, spleen, and peritoneal cavity on day 2 after I.V. injection (Supplemental Figure S3A-C). However, injecting Clo before the surgery (0–2 days) resulted in the deterioration of the health condition after the operation. Therefore, Clo was I.V. injected at the end of the surgical procedure (0 week), which maintained the reduced number of macrophages for a week (1-week Mø-depletion). Some mice received the second injection on day 7 to extend the period of macrophage depletion for another week (2-week Mø-depletion) (Figure 2(a)). Control mice alternatively received PBS liposome. The body weight of all operated mice gradually increased without a significant difference compared to the control (Supplemental Figure S4A). At 4 weeks post-surgery, the PBS hydrogel group showed minimal new bone formation (4.6%) in 2-week Mø-depleted mice (Figure 2(b) and (c)), whereas the healing rate without Clo treatment was approximately 22%. 14 This confirms the significant role of macrophages in the bone healing process.

Depletion of macrophages inhibits FGF18 augmented effect on calvarial bone healing.
The application of FGF18 + BMP2 hydrogel with PBS liposome injection (without Mø-depletion) resulted in new bone formation approximately 20.5% by 1 week post-surgery. The healing rate gradually increased to 64.2%, 79.1%, and 87.2% at 2, 3, and 4 weeks post-surgery, respectively, (Figure 2(b) and (c)) consistent with the previous report. 14 In 1-week Mø-depleted mice with the FGF18 + BMP2 hydrogel, the healing percentage at 1 and 2 weeks post-surgery were 4.5% and 36.0%, respectively. The bone healing rate continued to improve and reached 80.9% at 4 weeks (Figure 2(b) and (c)), which was almost equivalent to the PBS liposome control. In contrast, 2-week Mø-depleted mice with the FGF18 + BMP2 hydrogel remarkably failed in healing. The bone healing at 1 week was only 0.7% and increased to only 19.7% by 2 weeks with a small volume of nascent bone. Subsequently, the average bone healing was 40.9% and 57.2% at 3 and 4 weeks, respectively, which was significantly lower than those in the PBS liposome or the 1-week Mø-depletion groups (Figure 2(b) and (c)).
Of note, the bone healing activity of the BMP2 hydrogel was not significantly affected by 2-week Mø-depletion. At 4 weeks post-surgery, the healing rate of the 2-week Mø-depleted mice with BMP2 hydrogel was 56.3% that was similar to when the PBS liposome was injected (58.6%) (Supplemental Figure S4B-C). In addition, it was comparable to the new bone formation in the FGF18 + BMP2 hydrogel application with 2-week Mø depletion (Figure 2(d)). These observations strongly suggested that FGF18 functions through macrophage modulation, M2-Mø polarization, to stabilize BMP2-induced calvarial bone healing.
FGF18 is incapable of directly activating M2 macrophage polarization in bone marrow-derived macrophages
We next investigated how FGF18 induces M2-Mø polarization in vitro by using mouse-derived bone marrow (BM) cells (Supplemental Figure S1). First, we confirmed the differentiation of BM progenitor cells to non-polarized BM-derived macrophages (M0-Mø) by treating them with macrophage colony-stimulating factor (M-CSF) for 7 days. The high expression of two common mouse-specific macrophage markers, 22 F4/80 and CD11b, and the low expression of CD206 indicated the differentiation into M0-Mø (Supplemental Figure S1).
Since the transcripts of Fgfr1-3 were detected in both M0-Mø and BM progenitor cells (Supplemental Figure S5A), we first treated M0-Mø with FGF18 at 1 or 10 ng/ml (Figure 3(a)). Both conditions neither induced M2-Mø polarization nor altered the population of F4/80+CD11b+ cells (Figure 3(b), Supplemental Figure S5B). Prolonged treatment for 48 or 72 h did not show the polarization effects (Supplemental Figure S5C-D). As the positive control we applied interleukin-4 (IL-4) at 20 ng/ml for 24 h, which resulted in the robust M2-Mø polarization confirmed by the expression of F4/80, CD11b, and CD206 markers (Figure 3(b)). Notably, FGF18 application did not disrupt the proliferation of M0-Mø cells (Supplemental Figure S5E).

FGF18 is unable to directly induce M2 macrophage polarization in BM-derived macrophages.
We further investigated with RAW264.7 cells (a mouse macrophage cell line) to determine whether the inability of FGF18 to induce M2-Mø polarization depends on cell types. FGF18 treatment did not induce M2-Mø polarization (Supplemental Figure S5G). In addition, the proliferation and migration of RAW264.7 cells were not affected by FGF18 treatment (Supplemental Figure S5F, H-I).
We then added FGF18 to freshly isolated BM progenitor cells cultured with M-CSF to induce the differentiation of M0-Mø from day 0 (Figure 3(c)). After 7 days of culture, the FGF18-treated cells exhibited a comparable percentage of F4/80+CD11b+ cells (97.3-98.4%) compared to the control group (98.3%) (Figure 3(d)). However, the proportion of CD206+ M2-Mø within the F4/80+CD11b+ cell population did not show a significant difference from the control group (Figure 3(e)). In contrast, IL-4 treatment resulted in the induction of F4/80+CD11b+ cells at 95.6% and moderate M2-Mø polarization (Figure 3(d) and (e)). The failure of FGF18 to activate the expression of M2-Mø marker genes, Mrc1, Arg1, and resistin-like alpha (Retnla or Fizz1), (Figure 3(f)) suggested that FGF18 does not directly induce M2-Mø polarization in both M0-Mø and BM progenitor cells.
FGF18 activates M2 macrophage polarization in the total bone marrow cell culture
We then applied FGF18 to mouse total BM (tBM) cells, which includes both BM progenitor cells and BM stromal cells (BMSCs), to investigate the involvement of cellular interactions within the BM tissue. The tBM population was cultured for 7 days in the presence of M-CSF to induce M0-Mø differentiation. FGF18 (1 or 10 ng/ml) or IL-4 (20 ng/ml) was added either from day 0 or day 4, for 7-day or 3-day treatment, respectively (Figure 4(a)).

The addition of FGF18 to total BM cell culture increases M2 macrophage population.
After 7 days of tBM culture, immunofluorescent staining of F4/80 detected a consistent number of Mø cells (total M0-Mø/total cells) in all culture groups, ranging from 71.8% to 79.7% (Supplemental Figure S5J). This indicates that the differentiation into M0-Mø was not affected by FGF18 or IL-4 treatment. Among the total F4/80+ Mø cells, the CD206+ M2-Mø subpopulation was 26.4% in the control group (Figure 4(b) and (c)). Interestingly, the 7-day treatment with 1 or 10 ng/ml of FGF18 significantly increased the proportion of F4/80+CD206+ M2-Mø cells to 58.9% for 1 ng/ml and 67.1% for 10 ng/ml (Figure 4(b) and (c)). Similarly, the 3-day activation with FGF18 resulted in a notable elevation of F4/80+CD206+ M2-Mø cells to 58.6% with 1 ng/ml and 63.2% with 10 ng/ml (Figure 4(b) and (c)). The positive control group treated with IL-4 for 7 days and 3 days showed M2-Mø percentages of 82.7% and 75.8%, respectively (Figure 4(b) and (c)). RT-PCR analysis confirmed increased expression of Mrc1, Arg1, and Ym1 with both FGF18 and IL-4 treatments, while the upregulation of Fizz1 was only observed in the IL-4 groups (Figure 4(d)). Taken together, these results revealed that FGF18 activates M2-Mø polarization program by influencing other subpopulations in BM cells such as BMSCs.
FGF18 indirectly controls M2 macrophage polarization through bone marrow stromal cell activation
To investigate the factors responsible for inducing M2-Mø polarization, we cultured mouse BMSCs with 10 ng/ml of FGF18 for 72 h and collected the culture supernatant (BMSC-conditioned medium or BM-CM) (Supplemental Figure S6A). Subsequently, M0-Mø cells were cultured in the presence of BM-CM for 72 h. Notably, the original/non-filtered BM-CM (oBM-CM) led to higher levels of expression of Arg1 and Ym1 compared to the control BM-CM without FGF18 treatment (Figure 5(b) and (c)). This suggested that secreted factors from BMSCs are involved in the M2-Mø polarization.
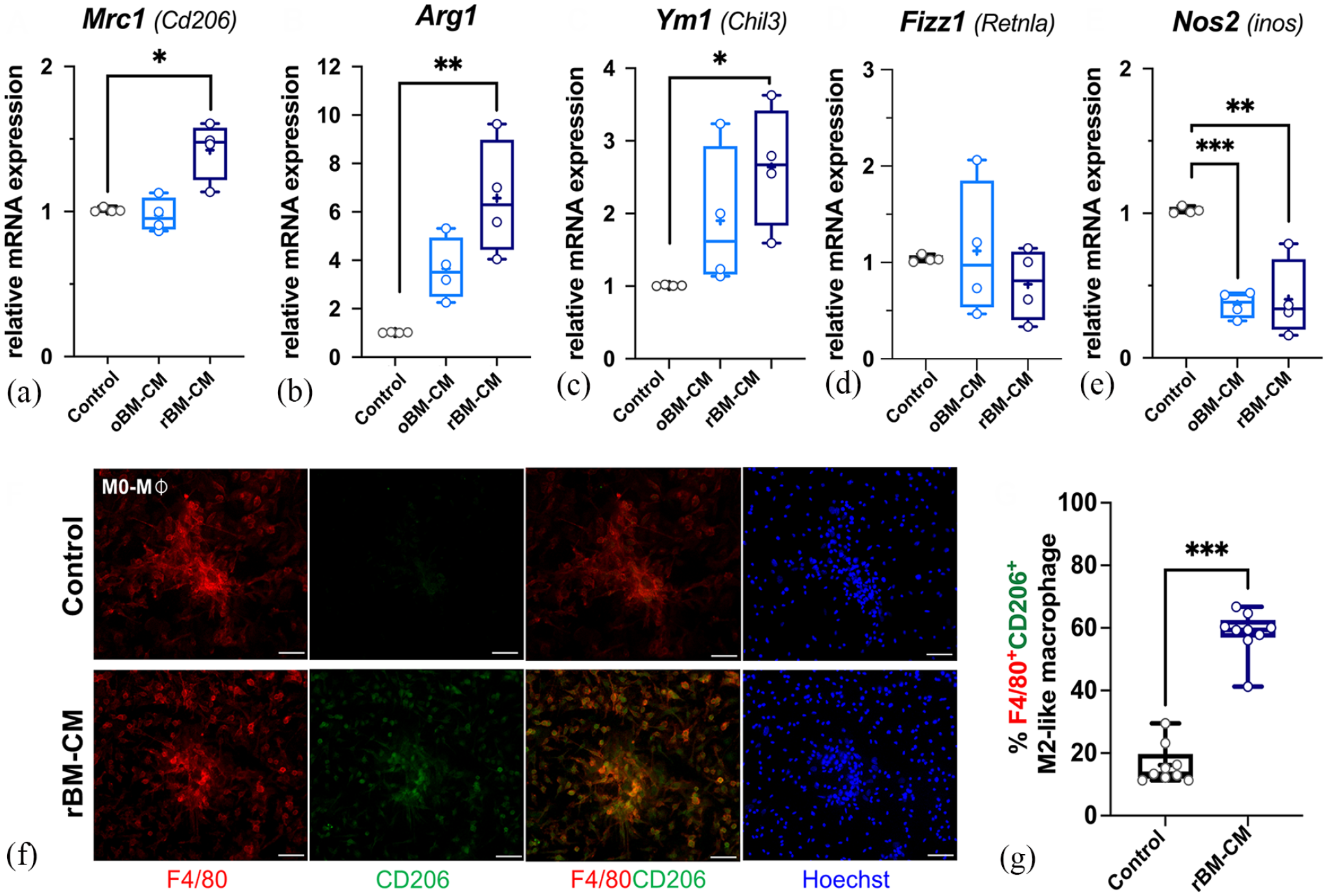
FGF18-treated BMSC-conditioned medium (BM-CM) stimulates M2 macrophage polarization.
The oBM-CM was next filtered to separate the secreting molecules at 10 kDa into two fractions: the residual (rBM-CM, ⩾10 kDa) and the filtered (fBM-CM, <10 kDa) fractions. Interestingly, the rBM-CM demonstrated a significant increase in the expression levels of Mrc1, Arg1, and Ym1 (Figure 5(a)–(c), Supplemental Figure S6B) as well as number of F4/80+CD206+ M2-Mø cells after 72 h (Figure 5(f) and (g)). In contrast, the fBM-CM hardly induced the expression of those marker genes (Supplemental Figure S6B). The expression of Fizz1 remained unchanged in all conditions (Figure 5(d), Supplemental Figure S6B). Of note, the transcription levels of Nos2, one of the M1-Mø markers, were notably downregulated in both oBM-CM and rBM-CM (Figure 5(e)). Thus, we hypothesized that FGF18 activates BMSCs to secrete unknown soluble mediators larger than 10 kDa, which play a role in promoting M2-Mø polarization.
FGF18-activated chemokine (c-c motif) ligand 2 (CCL2) derived from BMSC mediates in vitro M2 macrophage polarization
To explore the unknown secreted factors, we conducted microarray analyses using total RNA extracted from FGF18-treated BMSCs. The analysis focused on identifying upregulated genes with a fold-increase in expression value of two or more compared to untreated BMSCs (control). Next, we utilized gene ontology (GO) term analysis to narrow down upregulated genes based on their cellular component annotations, specifically “extracellular region (GO:0005576)” and “extracellular space (GO:0005615).” This filtering process resulted in the identification of 33 candidate genes (Supplemental Table S1).
Among them, we found chemokine (c-c motif) ligand 12 (Ccl12), Ccl2, Ccl9, Ccl7, and Ccl4, which have been implicated in the recruitment and activation of monocytes and macrophages. 36 Interestingly, the expression levels of well-known M2-Mø mediators, such as interleukin-4 (Il4), Il13, and Il10, remained unchanged (Figure 6(a), Supplemental Figure S7A-B). Additionally, several genes related to osteogenesis and bone matrix were also present in the gene list (Supplemental Figure S7C). Among the four most highly upregulated Ccl genes, chemokine ligand 2 (CCL2) has been exclusively associated with macrophage activation 37 and implicated in the inflammatory response during bone fracture healing and bone formation. 38

CCL2 derived from FGF18-stimulated BMSC mediates M2 macrophage polarization.
The treatment of M0-Mø with recombinant murine CCL2 for 24 h resulted in a significant increase in the expression of Arg1, Ym1, and Mrc1, while the expression of the anti-inflammatory cytokine Il10, remained unaffected (Figure 6(b)–(e)). Similarly, the treatment with rBM-CM for 24 h also upregulated the expression of these genes (Figure 6(b)–(d)). To confirm this effect, the rBM-CM was pretreated with anti-CCL2 neutralizing IgG antibody at the concentration of 1 or 2 µg/ml. After 72 h of culture, the rBM-CMs pretreated with the neutralizing antibody exhibited a significant decrease in induction of Arg1 and Ym1 expression (Figure 6(f) and (g)). The expression levels of other anti-inflammatory markers such as Mrc1 and Il10 were not affected (Figure 6(h) and (i)). These results suggested that CCL2, produced by FGF18-treated BMSCs, acts as a promising secreted mediator to activate M2-Mø polarization program in M0-Mø.
Consistent with the in vitro results, the relative mRNA levels of Ccl2 and Ccl12 were markedly higher in the defects with the FGF18 + BMP2 hydrogel compared to those treated with the BMP2 hydrogel at day 5 post-surgery in vivo (Figure 6(j) and (k)). Moreover, the expression levels of M2-Mø mediator genes, Il4 and Tgfb, were not altered (Supplemental Figure S7D-E). The findings indicated that the role of FGF18 in calvarial defect healing involves macrophage activation via the upregulation of Ccl2 during the early stages of bone healing.
FGF18-augmented activity in BMP2-induced calvarial defect healing model was demolished in Ccr2-/- mice
Previous studies reported that M0-Mø express chemokine (c-c motif) receptor 2 (CCR2), a CCL2-specific chemokine receptor.37,39,40 We applied the FGF18 + BMP2 hydrogel to the parietal bone defect in Ccr2 -/- mice. The body weight of all operated Ccr2 -/- mice was not compromised (Supplemental Figure S8A). However, the average healing rate at 1 week post-surgery showed a reduction of -17.1% (Figure 6(l) and (m), Supplemental Figure S8B). Nonetheless, bone healing subsequently improved, and the average healing rate reached 5.1% at 2 weeks post-surgery (Figure 6(l) and (m)). The defects showed gradual healing, with average closure rate at 18.9% and 34.9% at 3 and 4 weeks post-surgery, respectively (Figure 6(l) and (m)), which was significantly lower than wild-type mice (Figure 2(b) and (c), control). At 8 weeks, the average bone healing rate reached 58.0% in Ccr2 -/- mice (Figure 6(m)), which was similar to the healing with the BMP2 hydrogel in wild-type mice. 14
Moreover, the Ccr2 -/- mice treated with PBS hydrogel exhibited healing rate up to 16.4% (Figure 6(m), Supplemental Figure S8C) at 4 weeks post-surgery, which was comparable to that observed in wild-type mice, despite the presence of osteolysis in Ccr2 -/- mice.
It appears that the absence of CCR2 does not mediate the activity of FGF18 in augmenting bone healing. Therefore, the stabilizing effect of FGF18 in BMP2-induced calvarial defect healing can be partially attributed to the CCL2/CCR2 signaling pathway (Figure 6(n)).
Considering that the CCL2/CCR2 signaling pathway is involved in osteoclastogenesis 41 and osteoclasts play a role in the bone healing process, 42 we investigated whether FGF18 treatment could induce osteoclast differentiation. Analysis of the microarray data showed no upregulation of receptor activator of nuclear factor kappa-B ligand (Rankl) expression in FGF18-treated BMSCs. Furthermore, in vitro experiments demonstrated that treatment with FGF18 at concentrations of 2 ng/ml or 20 ng/ml did not alter osteoclast formation (Supplemental Figure S9A-B). Interestingly, when FGF18 was added to a co-culture of BM-derived monocytes and calvarial osteoblasts at a concentration of 20 ng/ml, the osteoclast formation was rather inhibited (Supplemental Figure S9C-D).
Discussion
In this study, we have uncovered the potential mechanism by which FGF18 enhances BMP2-dependent calvarial bone healing. This enhancement occurs through the indirect modulation of macrophages toward an M2-Mø phenotype, involving one of the chemoattractant cytokines, CCL2. Although the involvement of the FGF signaling pathway in skeletogenesis and skeletal metabolism by functioning in skeletogenic cells is widely recognized, this study revealed the new function of FGF signaling pathway in bone healing by influencing non-skeletogenic cells.
Previous research has reported the roles of FGF18 in bone and cartilage, particularly in osteogenesis, chondrogenesis and their repair processes. 6 Mouse calvarium-derived osteoblasts express Fgf2, Fgf9, Fgf18, Fgfr1, Fgfr2, and Fgfr3. 43 Notably, the transcription levels of Fgf18 specifically increase during long bone healing, 44 and FGF18 also influences the potential for osteogenic differentiation in MSC culture.12,13 Interestingly, a non-critical-sized bony defect in the calvarium of Fgf18 +/- mice exhibited compromised healing. 43 These findings suggest that FGF18 mainly functions through the skeletogenic cells in those biological events. In contrast, FGF18 has been found to play important roles in non-skeletal tissues, including the lung, central nervous tissues, and cancers. 45 FGF18 treatment also improved the survival of type-2 alveolar epithelial cells and reduce oxidative stress and inflammation. 46 Our findings contribute to the understanding of a novel role of FGF18 in modulating M2-Mø polarization, potentially leading to an anti-inflammatory response not only in bone healing processes but also in other biological processes.
The bone healing involves sequential steps, starting with inflammation and progressing to tissue remodeling. 47 Immune cell modulation has gained interest as a supplementary factor in bone healing by regulating the inflammatory conditions. The recruitment of appropriate immune and osteogenic cells to the site of injury is a critical event. 48 Bone-resident macrophages also play a crucial role in promoting osteoblast differentiation, mineralization, bone remodeling, and intramembranous bone repair.49,50 M2-Mø polarization has been shown to contribute to calvarial bone regeneration,23,51 which is consistent with our observation. Moreover, M2-Mø enhance osteogenic-related gene expression and mineralization in MSCs by regulating gene expression of Tgfb, Vegf as well as increasing BMP2 synthesis during bone healing process.52–54 Macrophages contribute to both intramembranous and endochondral ossification, and their depletion by clodronate liposome leads to incomplete long bone healing. 55 Similarly, depletion of macrophages during the early period of calvarial bone healing (1–2 weeks) resulted in impaired healing activity augmented by FGF18 supplement. In addition, the presence of CD206+ M2-Mø and increased ALP activity in the regenerating area at 1 week after surgery in the FGF18 + BMP2 hydrogel group suggested the important role of M2-Mø in bone healing and repair. This specific benefit provided by FGF18 in the FGF18 + BMP2 hydrogel might be setting up a favorable microenvironment for better healing at the early stage rather than creating sustained impact during all phases of calvarial bone healing. Indeed, our previous study expects that the maximal release of the growth factors from the CHPOA NanoClik hydrogel lasted for 10 days after the surgery.
The involvement of FGF/FGFR signaling pathway in inflammation, particularly in regulating the polarization of M1 and M2 macrophages have been demonstrated in various studies.25–27,56 It is clear that different FGF ligands exert distinct effects on the response of macrophage activation. In the Fgf2-deleted mice, M0-Mø predominantly polarized to an M1-like phenotype. 25 After FGF9 treatment in myocardium-infarcted diabetic mice, the number of CD206+ M2-Mø and expression levels of anti-inflammatory mediators, IL-10 and IL1-RA, increased. 26 FGF21/FGFR1 signaling was found to suppress nuclear factor-kappa B and enhance PPARγ activity in microglia, effectively inhibiting M1-like macrophage polarization in mice. 27 While the promotion of M2 polarization of macrophages through FGF signaling, particularly FGF2, has been widely studied in the field of cancer and tumor-associated macrophages, 57 its effect in tissue healing or regeneration processes remains elusive. In contrast, FGF23 stimulated an M1-like phenotype by elevating TNF-⍺ production. 56 FGF18 did not directly influence macrophage proliferation, differentiation, polarization, or migration in this study, despite the expression of Fgfr1-3 in macrophage progenitor cells and M0-Mø. The canonical FGF pathway activates four intracellular cascades such as PI3K-AKT, and STAT1, 3, 5.6,24 It would be interesting to investigate how the interplay between FGF ligands and receptors affects macrophage modulation in different biological contexts such as the treatment with combined FGF ligands.
Understanding the M2-Mø polarization pathway in wound healing remains challenging, although the well-established IL-4/IL-13 paradigm has been described based on in vitro studies. Accumulating data highlight the complexity of macrophage biology in different tissues. In our study, we observed an increased number of M2-Mø in the defect treated with the FGF18 + BMP2 hydrogel without upregulation of Il4, Il10, and Tgfb. Similarly, FGF18 treatment of BMSCs did not significantly affect the expression of those M2-Mø mediator genes after 24 h. A previous research reported that macrophage populations isolated from mouse dorsal skin wound tissues showed expression of Mrc1, Arg1, and Ym1 genes in the absence of IL-4/IL-13 and STAT6 upregulation, which are crucial signaling molecules for M2-Mø activation. 58 However, there is a possibility that the spatiotemporal timing was not appropriate to detect the expression of those molecules in our study. Especially, IL-4 is highly effective and a small amount of its expression could be sufficient to induce M2-Mø polarization. Although it is not significant, we observed that the expression of M2-Mø mediator Il10 is increased in the FGF18 + BMP2 hydrogel group after 5 days of operation in vivo.
Since CCL2 can stimulate not only migration of macrophages but also the induction of M2-Mø polarization, CCL2 secreted from FGF18-treated BMSCs may assist the M2-Mø polarization. There might be the crosstalk between the CCR2 and IL-4/IL-13/IL-10 signaling pathways in macrophages, which could be mediated through the JAK/STAT cascades.59,60 The interplay between BMSCs and macrophages in regulating the production of CCL2, IL-4 and other macrophage related molecules and their effect on macrophage modulation is not yet fully understood. While it has been observed that IL-4-treated BMSCs conditioned medium promotes the anti-inflammatory state of lipopolysaccharide-treated macrophages in vitro, 61 our healing model requires further investigations to elucidate the complexity of the crosstalk.
M0-Mø in Ccr2 -/- mice tend to differentiate into an M1-like macrophages. 59 This phenotypic shift may contribute to an increased inflammatory response and bone resorption following calvarial defect formation. CCL2/CCR2 signaling pathway has been proposed to play a role in recruiting mesenchymal progenitors during the early stages of fracture healing and remodeling.62,63 It also influences the migration of immune cells and regulates the polarization process of M1-Mø and M2-Mø during inflammation, although these processes are context-dependent.36,37,64 While the recruitment of those cells may be impaired in Ccr2 -/- mice, other chemokine signaling pathways are expected to remain functional and support the healing process. 36 The chemoattractant of monocytes is also mediated by CCL12, which binds to CCR2. 60 Therefore, the results in Ccr2 -/- mice may not solely reflect the action of CCL2 but also other CCR2-specific ligands, including CCL12 and CCL7.36,37 In addition, microarray analysis revealed the upregulation of these chemokines in BMSCs after FGF18 treatment. Other signaling pathways, except for CCL2/CCR2, could also function to create the immunological conditions necessary for bone healing. Further secretome analyses will uncover additional unidentified secreted factors in BMSCs or defect tissues that may contribute to M2-Mø polarization.
Clinical treatment with higher dose of BMP2 is associated with several adverse side effects, including the post-operative inflammatory surge. 5 This evidence suggests that BMP2 could act as an immunomodulator. A previous report demonstrated that BMP2 treatment decreased the expression of M1-Mø phenotypic markers in RAW264.7 cells, 65 which is consistent with our in vivo results showing a slight downregulation of pro-inflammatory cytokine markers in the BMP2 hydrogel group. Macrophage depletion by clodronate liposome treatment had no effect on BMP2-induced calvarial bone healing, indicating that the main function of BMP2 in our bone healing model is not the regulation of the immune system. To support this finding, it is well-known that BMP2 directly induces osteogenic induction and differentiation process in osteoprogenitor cells, osteoblast and BMSCs. 66 Indeed, the amount of BMP2 used in the study resulted in a wide range of unstable healing rates, 14 which indicates that the dosage of BMP2 employed might have been insufficient to influence the immunological status. Therefore, the co-delivery of FGF18 with a lower dose of BMP2 has the potential to diminish the side effect issues by stimulating M2-Mø polarization and promoting the formation of new bone with good quality, characterized by the presence of the marrow-like structure. 14
BMSCs are a non-hematopoietic subpopulation present in the bone marrow, capable of differentiating into various cell lineages upon specific stimulation. 67 They acquire immunomodulatory potential when exposed to appropriate stimuli 68 and release numerous factors such as prostaglandin E2, TGF-β, indoleamine 2,3-dioxygenase, CCL2, CXCL12, and extracellular vesicles.69,70 Bone marrow cavities were found in the parietal bone of operated mice, which could serve as a niche for BMSCs. It is intriguing to explore that other cranial cell populations, apart from BMSCs, could be involved in macrophage modulation in response to FGF18 in vivo. Recent studies have suggested that cranial suture stem cells, dura mater, and periosteum could serve as potential cell sources for calvarial tissue regeneration.71,72 Since bone healing potential declines with age, it is important to study the effectiveness of bone healing using the FGF18 + BMP2 hydrogel in old mice. While M2 macrophages are known to promote vascular formation through angiogenesis, 73 further investigations are warranted to understand the contribution of FGF/FGFR signaling pathways in calvarial bone repair, specifically in relation to other non-skeletal tissues in the cranium.
In conclusion, our data demonstrate the immunomodulatory activity of FGF18 in the context of BMP2-dependent osteogenic activity during mouse calvarial bone healing. Stimulation of BMSCs with FGF18 leads to the upregulation of specific chemokines, including CCL2, which contributes to the process of M2-Mø polarization. The control of macrophage activation by FGF18 offers several advantages over the single administration of BMP2 in both the quality and quantity of newly formed bone. Thus, these findings expand the potential auxiliary application of FGF18 in bone tissue engineering. Such advances hold promise for the development of novel strategies in future clinical settings for the treatment of craniofacial bone defects.
Supplemental Material
sj-docx-1-tej-10.1177_20417314231187960 – Supplemental material for Augmented effect of fibroblast growth factor 18 in bone morphogenetic protein 2-induced calvarial bone healing byactivation of CCL2/CCR2 axis onM2 macrophage polarization
Supplemental material, sj-docx-1-tej-10.1177_20417314231187960 for Augmented effect of fibroblast growth factor 18 in bone morphogenetic protein 2-induced calvarial bone healing byactivation of CCL2/CCR2 axis onM2 macrophage polarization by Worachat Namangkalakul, Shigenori Nagai, Chengxue Jin, Ken-ichi Nakahama, Yuki Yoshimoto, Satoshi Ueha, Kazunari Akiyoshi, Kouji Matsushima, Tomoki Nakashima, Masaki Takechi and Sachiko Iseki in Journal of Tissue Engineering
Footnotes
Acknowledgements
Authors thank Masako Akiyama for advice on statistical analysis. We also thank members of Department of Molecular Craniofacial Embryology and Oral Histology for their technical support and helpful assistance, Dr. Arun Kumar Rajendran for pilot experiments.
Author contributions
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Authors acknowledge funding from grants-in-aid from Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT) to Sachiko Iseki (17H04357 and 21H03098).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.