
Abstract
The tissue-engineered epidermal (TEE), composed of biocompatible vectors and autogenous functional cells, is a novel strategy to solve the problem of shortage of donor skin sources. The human primary keratinocyte (HPK), the major skin components, are self-evident vital in wound healing and was considered as one of the preferred seed cells for TEEs. Since the process of separating HPKs from the skin triggers a stress state of the cells, achieving its rapid adhesion and proliferation on biomaterials remains challenging. The key to the clinical application is to ensure the normal function of cells while improving the proliferation ability in vitro, and to complete the complex mesenchymal epithelialization to achieve tissue remodeling after vivo implantation. Herein, in order to aid HPKs adhesion and proliferation in vitro and promoting wound healing, we developed a three dimensional collagen scaffold with Y-27632 sustainedly released from the nanoplatform, hollow mesoporous organosilica nanoparticles (HMON). The results showed that the porous structure within the TEE supports the implanted HPKs expanding in a three-dimensional mode to jointly construct the tissue-engineered epidermis in vitro and inhibited the mitochondria-mediated cell apoptosis. It was confirmed that the TEEs with suitable degradation rate could maintain drug release after implantation and could accelerate vascularization of wound base and further revealed the involvement of mesenchymal transformation of transplanted HPKs during skin regeneration in a nude mouse model with full-thickness skin resection. In conclusion, our study highlights the great potential of constructing TEE using a nanoparticle platform for the treatment of large-area skin defects.
Keywords
Introduction
As the largest organ of the human body, the skin is the outer body barrier and plays a vital role in protecting the body, enabling tactile sensation, and regulating body temperature. 1 Severe wounds lead to a breach of the skin barrier, causing a massive loss of water electrolytes, which may result in acid-base imbalance, immune system disorders, and even a shock crisis. 2 Wound repair is a complex process affected by multiple factors. 3 As early as 4500 years ago, it was found that skin dressing or grafting for wound intervention may promote healing. 4 However, in practice, the skin of patients cannot be rapidly repaired by the graft, or tolerate complicated operations in such a poor condition. Furthermore, in the early stage after transplantation, severe wound contraction combined with a low degree of vascularization, 5 as well as aggregation of inflammatory factors 6 are the primary causes of skin graft necrosis. In the following decades, relevant studies on tissue-engineered skin replacement have rapidly developed. 7
Tissue-engineered epidermis (TEE) refers to expanding seed cells from the skin, and seeding them into a biodegradable bioscaffold in vitro for transplantation into a wound. In the rapid development of clinical applications,8,9 TEE, as the earliest attempt in tissue engineering, was most successfully applied to wound repair, especially for promoting the growth of basal tissue. 10 However, tissue engineering is rarely used in clinical practice. One reason is immunological rejection. Another reason is that the complexity of autologous cell differentiation or inverse differentiation determines the duration of tissue engineering construction for autologous organoids, which cannot support the conditions of patients with large skin loss.11,12
Apoptosis is a tightly controlled mode of programmed cell death, and mitochondrial apoptosis is the major form of regulated cell death. 13 Briefly, apoptosis is triggered when members of the proapoptotic family with a borane domain, such as BAD, BID, and BIM, are activated. The proapoptotic proteins BAX and BAK subsequently induce changes in mitochondrial outer membrane permeability, resulting in the release of cytochrome C from the intermembrane space of organelles. Then, cytochrome C forms an apoptotic body complex with Apaf-1 to activate caspase-9 and ultimately induce apoptosis. The successful construction of TEE requires a high survival rate of the implanted cells, which subsequently correlates with the efficacy of in vivo implantation. 14 Therefore, it is essential to explore new tools to downregulate apoptotic pathways within exogenous cells to improve survival and shorten the EET construction period.
RHO/ROCK signaling is known to play a key role in cell proliferation, differentiation, and apoptosis induced by the phosphorylation and downregulation of myosin phosphatase to increase actomyosin-based contractility. 15 As a potent inhibitor of ROCK, Y-27632 [(R)-(+)-trans-4-(1-Aminoethyl)-N-(4-pyridyl) cyclohexane carboxamide dihydrochloride] competitively binds to catalytic residues to inhibit ROCK. 16 Dakic et al. 17 demonstrated that Y-27632 cooperates with MYC without affecting the hTERT and P16/pRB pathways, while downregulating myc-mediated membrane vesicles and apoptotic responses by inhibiting the P53 pathway to immortalize primary keratinocytes. Y-27632 also promotes the evasion of induced pluripotent stem cells from dissociation-mediated apoptosis by inhibiting ROCK/MYOSIN signaling. 16
In this study, hollow mesoporous organosilica nanoparticles (HMON) were used to build a drug-carrying nanoparticle platform to resist the apoptosis of isolated cells in vivo and to build a 3-D TEE. Relying on the main source of skin grafts in clinical practice, we first used human primary keratinocytes (HPKs) isolated from the human scalp as the seed cells. Scaffolds with specialized pore structures display suitable mechanical properties to achieve the oxygen supply conditions required for biodegradation and cell growth. Interestingly, the scaffold sustained Y-27632 release maintained HPK proliferation and down-regulated mitochondrial-associated apoptotic pathways. Finally, the rapid proliferation of isolated HPKs was conducive to constructing the TEE and continued to exert anti-apoptotic effects on the implantation wound. Our approach represents a novel bioengineering strategy that is expected to have significant potential for future clinical translation.
Materials and methods
Synthesis of HMON
The synthesis of high-density silica (dSiO2) nanoparticles was based on the Stober process. 18 After 714 mL of ethanol, 10 mL of water, and 1.6 mL of ammonia were stirred for 10 min at room temperature, 2 mL of TEOS was added. The mixture was rotated for 1 h to collect dSiO2 nanoparticles, washed twice, and suspended in 20 mL of water. CTAC (8 g) and TEA (80 mg) were added to 200 mL of water and mixed thoroughly by stirring at a constant temperature of 50°C. The 20 mL dSiO2 solution described above was added and stirred at 80°C for 1 h, then 1.2 mL of TEOS was added and rotated for 1 h. The mixture was cooled to 50°C and stirred for 30 min, while the interior was specifically etched with 2.55 g sodium carbonate to obtain HMON. The resultant HMON was separated by centrifugation thrice (12,000 rpm, 10 min) and CTAC was extracted using NaCl2/methanol solution (1 wt%) for 24 h at room temperature. The HMON was dispersed in water.
Synthesis and characterization of Y@HMON
A total of 20 mg Y-27632 and 50 mg of HMON were mixed in ethanol solution and stirred for 24 h at room temperature to load Y-27632. The resultant Y@HMON was isolated by centrifugation (12,000 rpm, 10 min) thrice with an ethanol solution. The sustained-release capacity of Y-27632 was determined by measuring the concentration of Y@HMON over predetermined periods using a UV-visible spectrophotometer (Shimadzu, UV-2600 UV-vis, JPN). The chemical status and composition were determined using a Fourier Transform Infrared (FTIR) spectrometer and X-ray optoelectronic energy spectroscopy (XPS; ESCALAB250Xi, Thermo Fisher, USA). The diameter and zeta potential were measured using dynamic light scattering (DLS). Transmission electron microscopy (TEM) and elemental mapping images were obtained using a Hitachi HT7700 TEM (FEI, Hillsboro, OR, USA) system.
Fabrication of 3-D-CS/Y@HMON
A 1% w/v collagen solution was prepared by dissolving type I collagen powder from vituline tendon, provided by Lando Biomaterials Research & D Center (Shenzhen, China), in acetic acid (2% v/v, diluted in distilled water) at 4°C overnight. The pH was then adjusted to 7.4 with 1 N sodium hydroxide, and the collagen solution was then diluted to 0.5% with DMEM. Similarly, an alginate solution (5% w/v) was prepared by dissolving sodium alginate powder in a sterile, calcium-free phosphate buffer solution (PBS 1X). To create the sample, the alginate and collagen solutions were combined with the Y@HMON solutions in a ratio of 10:10:1, and fully dispersed with slow stirring to obtain a viscous solution.
A 100 mM solution of calcium chloride was used to crosslink the gel. After selecting 30 kV, 2 W, and 6.36 μm isotropic voxel sizes for image collection, 3121 projections from 360° were recorded with an exposure length of 1.5 s each. High-resolution X-ray computed tomography (XCT; Zeiss, Versa 520, USA) was used to assess the porosity and aperture of the freeze-dried hydrogel (n = 3).
Subsequently, Avizo (9.7.0, Sirmo Fisher Scientific, USA) was used to evaluate the 3-D data for the reconstruction. To determine the pore diameter and porosity of the target sample, which only contained the field of view (9.5 mm × 9.5 mm × 1.5 mm), it was extracted and converted into a binary picture using the interactive thresholding approach. The outermost pores at the edges of the samples were excluded from the analysis. A single layer of the collagen film was comprehensively compressed after full drying with a freeze-dryer to control its thickness from 0.2 to 0.3 mm.
The sample was weighed once and then placed in a shaking incubator (37°C, 150 rpm) soaked in 1 mL PBS, which was changed every 2 days. After removal from the buffer, the hydrogels were freeze-dried and weighed every 2 days. The amount of in vitro degradation was determined using the following formula (1):

where W1 is the initial weight of the material and W2 is the weight of the material after the degradation experiment. Each sample was mounted on an aluminum platform, and a layer of gold was sputtered on each sample under vacuum. The microstructures were observed using scanning electron microscopy (SEM; Sigma300, Zeiss).
Preparation of HPKs
In accordance with the Declaration of Helsinki Principles, ethical approval was granted by the Institute of Ethical Committee of Nanfang Hospital, Southern Medical School (approval number: NFEC-2021-265). HPKs were derived from the scalp tissues of 20 to 30-year-old men or women after dermatoplasty. Personal identity information was not obtained. To facilitate enzymatic lysis, the skin was cut into pieces and incubated overnight in dispase II (Roche, Mannheim, Germany) at 4°C. The next day, the epidermis was digested with 0.25% trypsin-EDTA solution (Coolaber, China) for 20 min at 37°C, as previously described. 19 The isolated keratinocytes were cultured in defined keratinocyte-SFM (ScienCell, 2111, USA) plus 20 μM Y-27632 (APExBIO, B1293, Houston, USA), 20 and the medium was changed every 3 days.
Construction of TEE
A schematic representation of the TEE production is shown in Scheme 1.
(1) After the scaffold was trimmed to a size of 1 × 1 cm and the matrix was stabilized on day 4, 3-D-CS and 3-D-CS/Y@HMON were placed under UV-C light for sterilization. The membranes were immersed in a defined KERATINOCYTE-SFM solution for 24 h.
(2) The previously prepared HPKs were added to the scaffold surface. A total of 106/mL keratinocytes in 100 μL defined keratinocyte-SFM per insert were directly seeded onto the formed sterile scaffold and then incubated at 37°C (with medium just covering the cells) to ensure that the keratinocytes could fully adhere. Cells attached to the substrate to generate a confluent cellular monolayer that is required for TEE preparation. The medium was constantly replaced to maintain a medium without floating TEE.

Schematic illustration of the construction of 3D-CS/Y@HMON-TEE and the mechanism of ameliorating the apoptosis of the implanted cells to repair diabetic wounds.
Cytotoxicity assay
Cell cytotoxicity was evaluated using the Cell Counting Kit-8 (CCK-8) assay, lactate dehydrogenase (LDH), and cell live/dead viability assays. Briefly, HPKs were seeded in 96-well plates and co-cultured overnight with 3-D-CS/Y@HMON. After 48 h of incubation, cell viability was evaluated based on the optical density (OD) value using CCK-8 (APExBIO, K1018). The LDH Cytotoxicity Assay Kit (BioVision, Milpitas, CA, USA) was used to determine the amount of LDH produced in the growth media, as previously described. 21 Cells were seeded in six-well plates at a density of 3 × 105 cells per well and co-cultured with 3-D-CS/Y@HMON. Triplicates of the sample solutions, 50 µL of each supernatant, and equal volumes of the reconstituted substrate were prepared in a 96-well plate. The plate was then read at 490 nm using an xMark microplate absorbance spectrophotometer after incubation for 15–20 min at room temperature in the dark until a yellow color emerged (Bio-Rad, Mississauga, ON, Canada). LDH levels were measured in cells treated with 3-D-CS/Y@HMON and 3-D-CS, with untreated cells serving as the negative control. The extent of LDH activity was calculated using equation (2):

A live/dead viability/cytotoxicity kit (Beyotime, L3224, Shanghai, China) was used to measure cell viability. Using the ImageJ software, the number of live (green fluorescence) and dead (red fluorescence) cells was calculated using an inverted fluorescence microscope (Olympus, Tokyo, Japan). The cell viability was estimated by dividing the number of viable cells by the total cell number. The average number of cells was recorded in at least three independent trials.
Effect of 3-D-CS/Y@HMON on HPKs in vitro
Cell proliferation was assessed utilizing the traditional MTT technique. A total of 104 PKS cells per well were seeded in six-well plates and co-cultured with 3-D-CS/Y@HMON for days, as previously described.22,23 MTT (Sigma-Aldrich, M5655-500MG, Ontario, Canada) was applied at one-tenth dilutions, and the cells were then incubated for 3 h at 37°C in the dark. A 0.05 N HCl-isopropanol solution (1 mL) was used to solubilize formazan crystals. Then, using an xMark reader, 4 × 200 µL of lysis buffer was transferred to a 96-well microplate (Bio-Rad, Mississauga, ON, Canada) to detect the absorbance at 550 nm. The percentage of proliferation in living cells was calculated using the following equation (3):

Detection of intracellular reactive oxygen species (ROS)
After co-culture with 3-D-CS/Y@HMON for 48 h, an ROS test kit (Solarbio, D6470, Beijing, China) was used for the dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay. Cells were allowed to react with the DCFH-DA probe at a final concentration of 10 μm for 30 min in the dark. The fluorescence signal of cells was captured at the same real-time exposure time (λex/λem = 488/520 nm). Fluorescence images were captured using an inverted fluorescence microscope.
Cell cycle analysis and apoptosis assays
To analyze the cell cycle or perform apoptosis assays, the HPKs treated with 3-D-CS/Y@HMON and 3-D-CS for 48 h were fixed overnight with 70% ethanol, stained for 30 min at 37°C in the dark with propidium iodide (PI; Beyotime Biotechnology, Shanghai, China), or with Annexin V-FITC Apoptosis Detection Kit (Solarbio, CA1020), and then examined using BD Biosciences FACSCalibur flow cytometry. 24
Cytobiology in TEE
TEE were cultured in a 12-well plate at 37°C, as described in 2.5. The specimens from each group were removed from the incubator on day 7 of culture. First, specimens were washed twice with preheated PBS and fixed with 4% paraformaldehyde for 30 min. Next, the fixative was washed away thrice with PBS at room temperature. For cell permeabilization, 0.2% Triton X-100 (Sigma-Aldrich, Ireland) was used in the proper sequence, followed by blocking with 1% bovine serum albumin (Sigma-Aldrich). Phalloidin (Bioss, C8013, Pek, CHN) and 4′,6-diamidino-2-phenylindole (DAPI; Abcam, ab285390) were used to stain the cytoskeleton and nuclei in the dark, respectively. After all preparations were completed, images of the cell morphology and adhesion of each specimen were recorded using laser scanning confocal microscopy (LSCM; 980, Zeiss). The cell distribution within the TEE was observed using a 3-D layer scanning function.
Evaluation of mitochondrial function
3-D-CS/Y@HMON and 3-D-CS were co-cultured with HPKs. Cells were collected 24 h after treatment to assess the MMPs and mitochondrial permeability transition pore (mPTP) using JC-1 (Solarbio, J8030) and mPTP kits (BestBio, BB-48122, Shanghai, China). In the mitochondria, JC-1 accumulates in a potential-dependent manner. Normal mitochondria have a rather high membrane potential, and JC-1 multimeric aggregates in the mitochondrial matrix showed red fluorescence. Defective mitochondria did not allow the accumulation of JC-1. The matrix released green fluorescence as a result of a drop in or loss of the MMP. The cells were treated with PBS twice before staining with JC-1 (5 μg/mL) for 15 min, mixed, and cultured for 20 min at room temperature without exposure to light. JC-1 staining buffer was used to remove excess staining solution. For mPTP staining, the CAI staining working solution was diluted with PBS at a ratio of 1:100 and then incubated with the cells for 5 min or until the entire sheet of green material was used to determine the opening of the mitochondrial membrane channels. 5 µL of quencher and 200 μL Hoechst 33342 (ab228551) were then added, and incubation was maintained for another 25–30 min for microscopic inspection. LSCM was used to obtain the fluorescence images.
Prtreated-HPKs were collected to evaluate mitochondrial active oxygen. These cells were then treated with 5 μM MitoSOX Red for 30 min at 37°C and washed twice with PBS (MitoSOX; M36008, ThermoFisher). After staining with Hoechst 33342 (GC63515) for 15 min, the images were captured using LSCM. For parallel experiments, cells were washed with PBS for flow cytometric immunofluorescence analysis measured by a Flow Cytometer (FACSVerse, USA).
Western blotting
Treated cells and skin samples were collected for total protein extraction. Proteins were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to PVDF membranes (Millipore, Bedford, Germany). The PVDF membranes were treated with a specific primary antibody for 4 h at 4°C, followed by blocking with 5% BSA. After 12 h, membranes were incubated with secondary antibodies (1:20,000) for 1 h. Finally, a Tanon-5200 (Tanon, Shanghai, China) was used to capture the protein bands. The primary antibodies used were: human Bax antibody (1:1000; Abcam, ab32503), human Bcl-2 antibody (1:1000; Abcam, ab32124), human caspase 3 antibody (1:1000; Abcam, ab13847), human caspase-9 antibody (1:1000; Abcam, ab32539), mouse anti-human glyceraldehyde-3-phosphate dehydrogenase antibody (GAPDH; 1:5000; Abcam, ab8245), anti-E-cadherin (1:5000; 20874-1-AP, Thermo Fisher), anti-vimentin (1:1000; 10366-1-AP, Thermo Fisher), and anti-fibronectin (1:1000; 15613-1-AP, Thermo Fisher).
Wound healing assay in a full-thickness skin model in vivo
This study was approved by the Animal Experimentation Ethics Committee of Nanfang Hospital, Southern Medical School (approval number: IACUC-LAC-20221015-001). The principle of minimizing animal suffering and the number of rodents used were always followed in this study. BALA-C/nu mice (no sex limit, n = 36, weighing 20–24 g) were purchased from the Experimental Animal Centre of Southern Medical University and kept in a specific pathogen-free environment (22°C ±2°C) with free access to standard food and water. The experiment commenced after an 1-week adaptation to the environment, and the animals were randomly divided into four groups (n = 9/group): sham (blank control), 3-D-CS, 3-D-CS-TEE, and 3-D-CS/Y@HMON-TEE. First, mice underwent anesthesia in the induction chamber with isoflurane (3% in O2 at 0.6 L/min). Then, they were immediately transferred to an experimental heating pad set at 37°C with the limbs stretched. During the course of the experiment, general anesthesia was maintained using a nose cone that could sustainably release the gas flow (3% in O2 at 0.3 L/min). The dorsal skin along both sides of the spine was excised in full thickness without the musculature beneath it being damaged using ophthalmic scissors under sterile surgical conditions to form a square area of approximately 1 cm2, which was similar to the size of the graft. After bleeding was controlled with appropriate pressure, the wound was covered with the full-thickness TEE on day 1. The membrane was then sutured to the edge of the wound skin. Amicrobic Vaseline gauze and sterile dressing (REF# 1624w, 3 M Healthcare) were applied sequentially against the graft to the wound bed to prevent scratching and infection. Dressing fixation should not limit the activity of the animals, including feeding in water and defecation. The mice were monitored daily, with external dressings replaced every 2 days. A digital camera (iPhone 12 Pro) was used to photograph the wound sites on days 0, 7, 14, and 21. The resulting images were quantified using the ImageJ software.
Cell migration
Additional model mice (2 × 2 cm wounds) were prepared for in vivo spectrum imaging. Following the manufacturer’s instructions, 25 the HPKs were labeled with the cell membrane-labeling dye PKH-26 (Sigma-Aldrich) before grafting. After anesthesia, the mice were restrained in a rodent restrainer for TEE transplantation. Mice were monitored weekly using a spectrum in vivo imaging system (IVIS, AZ, USA).
The following primary antibodies were used: monoclonal mouse anti-human vimentin (cat. 3390, MA, USA) and DAPI. The primary antibodies were used for immunofluorescence staining overnight at 4°C. The stained tissues were imaged using an EVOS FL auto inverted fluorescence microscope.
Hematoxylin and eosin (H&E), Ki-67, and Masson’s trichrome staining
A 10% formalin solution (Sigma-Aldrich, Ireland) was used to fix the tissues taken from the grafted site. The tissues were then dehydrated using an ethanol wash before embedding in paraffin. A Leica RM2255 microtome was used to slice the implanted tissue into 5 μm pieces. H&E (Abcam, ab245880), Ki-67 (Abcam, ab15580), and Masson’s trichrome staining (Abcam, ab150669) were performed following dewaxing and rehydration using histological staining kits according to the manufacturer’s instructions. Collagen fibers were stained using Masson’s trichrome stain kit, and cell proliferation in tissues was determined by Ki-67 staining. Images were captured using a light microscope (ECLIPSE 90i; Nikon, Tokyo, Japan).
Enzyme-linked immunosorbent assay (ELISA)
TGF-β1 concentration was measured separately using ELISA (R&D Systems, Minneapolis, MN, USA; Thermo Scientific, USA). A 96-well microplate spectrophotometer (FLUOstar Omega, BMG LABTECH, USA) was used to measure absorbance at 450 nm. The former test samples contained three time points, whereas the latter only measured the concentration on day 7.
Reverse transcription-quantitative PCR
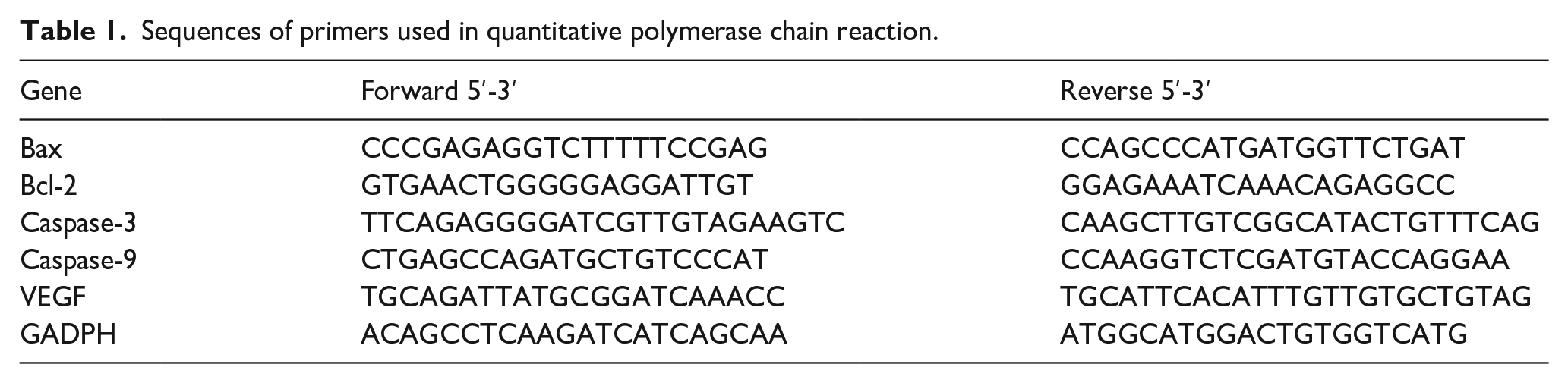
A fluorescence quantitative PCR kit (TaKaRa, Dalian, Liaoning, China) was used to assess the relative mRNA expression levels. The reaction mixture comprised 5.3 µL of 2 × Taq Master Mix, 1 µL of cDNA template, 1 µL of forward primer (5 μM), 1 µL of reverse primer (5 μM), and 11.7 µL of RNase-free H2O. Pre-denaturation at 95°C for 5 min, followed by 35 cycles of denaturation at 95°C, were the reaction conditions, followed by 45 s of retraction, 45 s of annealing at 56°C, and 45 s of extension at 72°C. Real-time fluorescence PCR was performed using GAPDH as an internal reference and ABI7500 equipment (Applied Biosystems, Inc., Carlsbad, CA, USA). The relative mRNA expression of the target gene was determined using the 2-ΔΔCt formula. The experiments were conducted in triplicate. For more details in Table 1.
Sequences of primers used in quantitative polymerase chain reaction.
Statistical analysis
Data from at least three independent experiments are presented as the mean ± standard deviation. GraphPad was used along with an unpaired Student’s t-test or one-way analysis of variance (ANOVA) to assess statistical significance in the experiments. Differences between two groups were analyzed using an unpaired Student’s t-test, while differences among three or more groups were analyzed using one-way ANOVA. A p-value of <0.05 indicated statistically significant differences.
Results and discussion
Preparation and characterization of 3-D-CS/Y@HMON
Previous studies have shown that silicon nanoparticles are widely used for drug delivery owing to their excellent loading capacity, biocompatibility, and biodegradability.26,27 In this study, we used a selective etching strategy to synthesize HMON as nanocarriers. First, uniformly dense silica (dSiO2) nanoparticles, approximately 180 nm in diameter, were synthesized as a template with negative surface charges (−15.3 ± 0.8 mV). As shown in Figure S1A, the signature band spectra around 1200 cm−l on the intense ~1090 cm−l TO band reflect the longitudinal optic modes. The band at ~460 cm−l was assigned to the bending mode of the siloxane linkages, and the Si-O stretch vibrations of the silanol (Si-OH) groups were observed at ~956 cm−l, indicating that HMON was successfully constructed. TEM images revealed that the HMON was spherical in shape (~180 nm in diameter). The elemental mapping images of HMON showed a homogenous distribution of Si and O (Figure 1(a)). The hydrodynamic diameter of the nanoparticles was stable after loading and modification, with a negative surface charge (−20.7 ± 1.2) mV (Figure 1(b)). As shown in Figure 1(c), the loading of Y-27632 did not affect the structure and morphology of the nanoparticles. XPS confirmed the presence of C (C 1s), O (O 1s), Si (Si 2p), and S (S 2p) in Y@HMON (Figure 1(d)).

Characterization. (a) TEM images and elemental mapping (Si, O, Merge) of Y@HMON. (b) Diameter distribution of Y@HMON and (c) Zeta potential of HMON and Y@HMON. (d) XPS images of HMON. (e) UV-vis absorption spectrum of Y-27632 (Blue), HMON (red), and Y@HMON (Black). (f) Cumulative release profiles of Y-27632 encapsulated in 3D-CS/Y and 3D-CS/Y@HMON. (g) Photograph images and 3-D scanning views of the internal structure using CLSM. (h) SEM images of 3D-CS, 3D-CS/Y@HMON, and Y@HMON.
We designed a collagen scaffold with a 3-D aperture structure of 0.2–0.3 mm thickness by referring to previous methods (3-D-CS). 28 From the UV-visible spectra shown in Figure 1(e), the Y@HMON nanoparticles had an absorption peak at ~269 nm, indicating that Y-27632 was successfully encapsulated into the HMON nanoparticles. The drug release time of 3-D-CS/Y@HMON was detected for up to ~120 h, whereas it was only detected for ~60 h for 3-D-CS directly mixed with Y-27632 (Figure 1(f)). Y@HMON was observed in the 3-D collagen scaffold. The image analysis in the XCT tomography and SEM images showed that the 3-D-CS had a highly interconnected porous structure (Figure 1(g) and (h)), with a total porosity of 50.27% ± 11.71%, and pore size ranging between 20 and 160 µm (Table 2; Figure S1B). The lyophilized weight was measured at different time points in PBS to assess degradation. Degradation occurred at a slow rate, with an overall weight decrease of 83% on day 14, compared to 97% on day 21 (Figure S1C). Based on this, we believe that 3-D-CS/Y@HMON was successfully prepared with the ability of sustained Y-27632 release, thus enabling the scaffold to exert long-term anti-apoptotic, pro-proliferative, and anti-inflammatory effects.
Porosity data and degradation time for crosslinked scaffold 3D-CS/Y@HMON. Data are reported as mean ± SD (n = 3).

The HPKs maintained cell proliferation capacity within the 3-D-CS/Y@HMON-TEE
As the main constituent cells of the epidermis, HPKs play an extremely important role in tissue engineering, skin construction, and wound cell treatment, but the disadvantages of low adherence rate, proliferation difficulty, and poor vitality during isolation in vitro hinder their practical application. Therefore, we examined if 3-D-CS/Y@HMON could support the proliferation of HPKs when cultured in vitro. HPKs were isolated from human scalp tissue and co-cultured using serum-free medium (DKSFM) with various materials. Compared to the 3-D-CS group without Y@HMON, the results in the 3-D-CS/Y@HMON group confirmed that the sustained release of Y-27632 from 3-D-CS/Y@HMON increased the adherence rate of HPKs (Figure 2(a) and (b)). On the one hand, compared to the 3-D-CS (4 N + S: ~17.79%) blank group, more cells were in the proliferative period in the presence of the nanoparticle platform (4 N + S: ~34.1%) as demonstrated using flow cytometry (Figure 2(c) and (d)). This finding was also confirmed by the MTT assay results (Figure 2(f)).

Effects of 3D-CS/Y@HMON on HPKs in vitro. (a) The adhesion on day 1 of HPK after 3D-CS and 3D-CS/Y@HMON added were observed under bright field, and (b) its quantification result. (c and d) Flow cytometry for cell cycle after treatment of 3D-CS and 3D-CS/Y@HMON. (e) LDH leakage rate, (f) MTT, and (i) CCK-8 assay at preset time points after 3D-CS and 3D-CS/Y@HMON treated. (g and h) Live and dead staining of HPK and HUVEC cells cultured with Y-27632 on 24 and 72 h. n = 3, n.s.: no significant, and ***p < 0.001.
In vitro cytotoxicity of 3-D-CS/Y@HMON
It is well known that biological-origin collagen material shows good biocompatibility and is non-immunogenic without being cytotoxic. 29 We did not observe any cytotoxic effects of 3-D-CS/Y@HMON within 36 h, as determined by both the CCK-8 and LDH assays (Figure 2(e) and (g)). The cell dead/live assay indicated that human umbilical vein endothelial cells (HUVEC) and HPKs showed no significant differences during the 72 h co-culture, regardless of the presence or absence of Y@HMON (Figure 2(h) and (i)). Overall, the collagen scaffold serves as a representation of the extracellular matrix (ECM), and is a source of biochemical and biophysical cues that support cell growth. These signals primarily improve primary cell adhesion and attachment as well as cell survivability and function. However, their long-term impact on cell proliferation remains unknown.
The nanoparticle platform successfully participated in the construction of highly active TEE substitutes
HPKs were seeded on 3-D-CS and 3-D-CS/Y@HMON to explore their migration and proliferation (described as 3-D-CS-TEE and 3-D-CS/Y@HMON-TEE). On day 6 after inoculation, HPKs in the 3-D-CS/Y@HMON group were more quickly expanding in a fusion state, and new epidermal tissue was constructed, as observed using LSCM (Figure 3(a)). Considering the presence of visual field blind spots in 2D image observations, we further explored the distribution of cells within the TEE. In the SEM images on day 3, the cells were attached to the porous structure of the CMDS, and some of the cells were connected to the collagen fibers in the form of cellular antennae (Figure 3(b)). Interestingly, compared to the previously lyophilized collagen scaffold (Figure 1(b)), the medium-soaked scaffold showed a certain degree of water absorption, but retained a 3-D porous structure, providing space for cells to embed and grow. This facilitates the large expansion of implantable cells in vitro, and the later implantation acts as a “cell bank,” ensuring that the wound epidermis is adequately covered by KCs and expanded to promote healing. The growth of HPKs inside TEE on day 3 was reduced using the 3-D layer scanning function of LSM 980 (Figure 3(c)). We found that the TEE with Y@HMON had a higher cell density, but both cell types were uniformly distributed without any morphological abnormalities.

Cells growth status inside the 3-D collagen scaffold. (a) Immunofluorescence staining (DAPI) of HPKs in TEEs. (b) SEM images of 3D-CS or 3D-CS/Y@HMON-associated HPKs. White arrows indicated cells attached on the collagen fiber. (c) 3-D view of TEEs growth on day 3 was constructed from z-stack slices through CLSM.
Anti-apoptosis effect of the nanoparticle platform in vitro
Subsequently, we explored the relevant mechanisms of the rapid construction of TEE using nanoparticle platforms. Previous studies have shown that Y-27632 can prevent cell death through the downregulation of rapid apoptosis caused by both extrinsic and intrinsic pathways.17,30 The isolation process from the body, as an external stimulus, will undoubtedly stress the human skin cells, as do the cells implanted within TEE, which will also produce a series of feedbacks because of biomechanical changes.
To explore the role of 3-D-CS/Y@HMON-TEE in improving apoptosis, 3-D-CS/Y@HMON loaded with a gradient concentration of Y-27632 (0–5 µM) were co-cultured stratified with HPKs for 48 h based on the previous UV-vis results shown in Figure 1(g). Flow cytometry analysis showed that the rate of apoptosis gradually decreased as the initial concentration of Y-27632 increased, indicating that the initial pre-loading concentration of 5 µM was optimal for improving apoptosis (Figure 4(a) and (b)). The anti-apoptotic properties of 3-D-CS/Y@HMON were confirmed. Meanwhile, the intracellular ROS levels of HPKs from different treatments were determined, and it was found that 3-D-CS/Y@HMON treatment could significantly downregulate the level of ROS produced by oxidative stress (Figure 4(c)). Quantitative analysis confirmed a 4.18-fold decrease in the ROS content in 3-D-CS/Y @HMSON-treated cells compared to that in 3-D-CS-treated cells (Figure 4(g)).
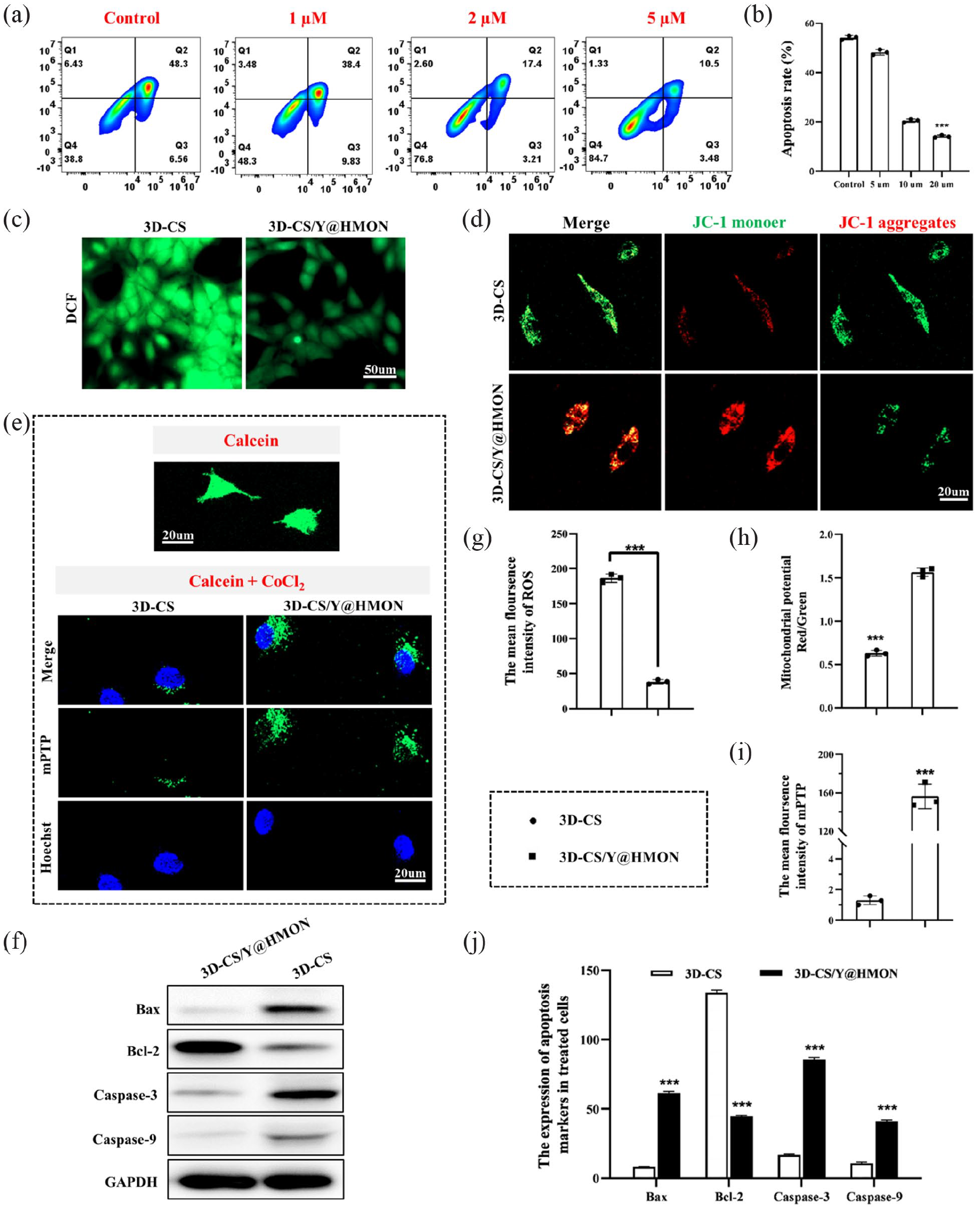
In vitro validation of mitigating HPKs apoptosis for co-cultured with 3D-CS/Y@HMON continuously releasing Y-27632. (a) Cells apoptosis under Y-27632 treated with gradient concentration was determined by flow cytometry, and (b) Statistics analyzed the apoptosis rate of HPKs. (c and g) Confocal laser scanning microscopy (CLSM) images of HPKs cells for the detection of intracellular ROS, before and after incubating within 3D-CS or 3D-CS/Y@HMON. Green: DCF fluorescence generated from the oxidization of nonfluorescent DCFH by ROS. (d) Mitochondrial membrane potential of cells, and (h) their corresponding JC-1 aggregates (red) and monomer (green) intensities. (e and i) CLSM images of mPTP in HPKs co-cultured with 3D-CS or 3D-CS/Y@HMON. Blue: cell nuclei stained by Hoechst 33342 dye; green: calcein fluorescence for the detection of intracellular mPTP before and after Cocl2-treated. (f and j) The quantity of Bax, bcl-2, caspase-9, caspase-3, and GAPDH proteins analyzed by Western blot in HPKs after incubation with 3D-CS or 3D-CS/Y@HMON. N = 3, ***p < 0.001.
Mitochondria-mediated apoptosis refers to apoptosis triggered by factors, such as DNA damage and growth factor lack. It causes Bad/Bak proteins (BCL-2 family members) to form an oligomer complex that is then inserted into the mitochondrial outer mode gap, resulting in mitochondrial membrane permeability change, transmembrane potential loss, and apoptosis-related factor release. 31 In contrast to the blank group 3-D-CS, 3D-CS/Y@HMON-treated HPKs showed lower levels of mitochondrial reactive oxygen species (mtROS) (Figure S2A-C), which preliminarily verified the anti-apoptotic hypothesis. Maintaining the normal potential of the mitochondrial membrane is the premise of normal mitochondrial function. As abnormal mitochondrial function is the beginning of gradual cell death, it is necessary to maintain normal mitochondrial oxidative phosphorylation. JC-1 staining showed strong red fluorescence (JC-1 aggregates) inside the mitochondria of HPKs co-cultured with 3-D-CS/Y@HMON, indicating that mitochondrial status was not affected. In contrast, drug-free 3-D-CS produced strong green fluorescence (JC-1 monomer) and weak red fluorescence, indicating that the mitochondrial membrane was destroyed and showed a high negative potential (Figure 4(d) and (h)). This was also accompanied by closure of the mPTP (Figure 4(e) and (i)). The mPTP plays a crucial role in cell physiology and pathology, and excessive opening is involved in the pathophysiology of human diseases by causing cellular and mitochondrial ion and oxidative imbalances. 32 It exhibits mitochondrial swelling that triggers the release of cytochrome C, impairs respiration, and ultimately causes cell death. 33 Calcein selectively accumulates within mitochondria, showing fluorescent staining, and fluorescence is quenched by CoCl2 when calcein escapes outside the mitochondria due to excessive opening of the mPTP. 34 In this way, we observed that the opening rate of the mPTP following treatment with 3-D-CS/Y@HMON was lower than that of 3-D-CS.
As ROS are known to induce apoptosis through Bcl-2 family proteins and caspase activation, Bcl-2 (anti-apoptotic protein), Bax (proapoptotic protein), and caspasase-3/9 were tested using western blotting to verify the ability of the Y-27632-loaded nanoparticle platform to reduce apoptosis. As shown in Figure 4(f), the nanoparticle platform loaded with Y-27632 upregulated Bcl-2, an anti-apoptosis-associated protein, and downregulated Bax, an apoptosis-associated protein (Figure 4(j)). The levels of Caspase-3 and Caspase-9, as the most important terminal proteases during apoptosis, were also reduced. These results strongly confirmed that the nanoparticle platform 3-D-CS/Y@HMON effectively downregulated the mitochondria-mediated apoptotic pathway, thus ensuring the viability of the implanted cells.
3-D-CS/Y@HMON-TEE markedly promoted wound healing efficacy
The mouse model of acute full-thickness wounds was used to assess the therapeutic potential of 3-D-CS/Y@HMON-TEE (Figure 5(a)). The control group that only received silicone film coverage was considered the baseline for self-healing, whereas plain 3-D-CS/Y@HMON without inoculated cell seeds served as a blank control. The wound areas were traced at specific time points after the TEE was placed in the skin removed area. From the wound photographs taken (Figure 5(b), the TEE repaired the wounds significantly faster than non-cell therapy, which indicated HPK contributes to the wound repair. Non-cell therapy without HPK implanted relies more on the early body’s skin contraction function and the migratory coverage of keratinocytes at the edge of the later wound than on the TEE direct repair mode. 30 This was also confirmed in Figure S3, where the 3D-CS/Y@HMON-TEE group showed a more ordered skin tissue structure compared to the 3D-CS/Y@HMON group. Quantification of the wound size at each time point yielded the average days of healing in each group: 3-D-CS/Y@HMON-TEE (9.4 ± 0.2 days), 3-D-CS-TEE (10.7 ± 0.3 days), 3-D-CS/Y@HMON (16.4 ± 0.4 days), and control (19.3 ± 0.5 days) (Figure 5(f)). As the healing-promoting effect of TEE has been clarified, we only discussed the advantages of the nanoparticle platform loaded with Y-27632.

3D-CS/Y@HMON-TEE enhances cells proliferation to promotes acute full-thickness wound healing in vivo. (a) Schematic illustration of animal experiments. (b) Photographs of infected wounds and (f) wound healing time for various groups on 0, 7, 14, and 21 days. Black triangle indicated the shedding of the nascent epidermis, Scale bar: 1 cm. (c) H&E staining of the wound skin tissue, (d) immunohistochemistry of Ki-67, and (g) its statistical analysis. Dashed circles indicated the anchor structures connecting the nascent epidermis to the basal tissue. (e and h) The collagen deposition at day 28 in the regenerated tissue was examined by Masson trichrome staining where the blue intensity showed the arrangement and the concentration of collagen deposition. n = 3, *p < 0.05, ***p < 0.001.
It should be noted that, although 3-D-CS-TEE enhanced wound healing compared to non-cell implantation therapy, shedding of the epidermis was observed by gently wiping the regenerated area with a wet cotton swab (shown as a black triangle in Figure 5(b)). In fact, the TEE implantation body cannot establish a close connection with the basal tissue, which was one of the difficulties in the clinical application of TEE transformation in previous studies. The H&E staining results showed that the wound epidermis treated with 3-D-CS/Y@HMON-TEE was thicker (Figure 5(c)), possibly associated with the cell proliferation capacity, as confirmed by the corresponding immunohistochemical Ki-67 staining (Figure 5(d); Figure S4). New epidermal layer cells on day 7 after implantation demonstrated a high degree of proliferative viability (Figure 5(g)), which advanced the wound healing process overall. On day 21, anchor-like structures appeared (shown as dashed circles in Figure 5(c)), indicating that the new tissue structure was stabilized. In addition, the residual collagen scaffold was seen on day 14 without local inflammation, but basic degradation occurred on day 21, which was consistent with the previous in vitro degradation experiments (Figure S5), which explained the biocompatibility of the material.
The subcutaneous collagen scaffold promotes basal granulation tissue repair in an orderly manner. We then assessed collagen deposition in the regenerated dermis using Masson’s trichrome staining (Figure 5(e)). Collagen deposition, represented in blue, was much more ordered and distributed in nascent tissues treated with 3-D-CS/Y@HMON-TEE. 35 To quantitatively analyze the collagen recovery, we calculated the collagen index (CI) in the histological images. 36 The data showed that the 3-D-CS/Y@HMON-TEE treatment (0.82 ± 0.16) significantly improved the collagen structure (Figure 5(h)), as compared to the 3-D-CS-TEE-treated CI (0.22 ± 0.07). Thus, compared with 3-D-CS-TEE without the nanoparticle platform, treatment with 3-D-CS/Y@HMON-TEE connected the epidermis more closely to the dermis, which was associated with the migration and growth of endothelial cells and fibroblasts near the wound in the stent. Thus, the use of 3-D-CS/Y@HMON-TEE effectively overcomes the hindrance of cracked epidermis and blisters in previous tissue engineering applications. The orderly collagen arrangement may be more conducive to the later scar formation, but the time course in this experiment was too short to avoid further discussion of the scar correlation.
HPKs undergo epithelial-to-mesenchymal transition with the aid of the nanoparticle platform
Wound healing is a dynamic process involving the interaction of various cells, cytokines, and ECM. To further reveal the cytobiology during nanoparticle platform-assisted wound repair, we collected skin tissue at the indicated time points and performed immunofluorescence analysis. After injury, functional keratinocytes around the wound site respond to potential cytokines in the local tissue, causing the loss of tight junctions and gaining the ability to migrate to the middle of the injury site. Influenced by the complex microenvironment of the wound, HPKs lose their epithelial characteristics and acquire the typical characteristics of mesenchymal cells and their migration ability. These processes are accompanied by complex changes in cell structure and behavior, defined as epithelial-mesenchymal transformation (EMT). Cell migration capacity is then enhanced to participate in normal or fibrotic repair after tissue trauma. 37 Therefore, to examine EMT-mediated wound repair, we stained tissue sections with fluorescently labeled anti-vimentin antibodies, and the results suggested that exogenous implanted cells are involved in re-epithelialization (Figure 6(a)). The antibodies used in the experiments did not cross-react with mouse proteins but only reacted with human proteins, indicating that the cells were derived from the seeds within the scaffold. The sustained increase in the vimentin expression levels from day 7 to day 21 in the 3-D-CS-TEE and 3-D-CS/Y@HMON-treated groups supported the functional role of transplanted keratinocytes in the construction of new tissues (Figure 6(b) and (c)).

The mesenchymal transformation of transplanted keratinocytes assisted wound healing in vivo. (a–c) The 3D-CS-TEE and 3D-CS/Y@HMON-TEE graft tissue were harvested at day 7, 14, and 21 followed by sectioning perpendicular to the dermis plane. The tissue was then stained against pan-CK (blue) and vimentin (green) and imaged with confocal microscopy. The representative data is shown. (d and f) Molecular changes associated with EMT: fibronectin, E-cadherin and vimentin analyzed by Western blot in vitro and in vivo. (e and g) Corresponding quantitative data from Western blot. (h) Concentration of TGF-β1 on day 7 and day 14. (i) The TEEs-carried HPKs were fluorescently labeled prior to grafting on mice. Mice was imaged on day 1 or day 7. The red arrow indicates cells migrating to the surrounding area. n = 3, n.s.: no significant, and ***p < 0.001.
Compared to the control group, consistent with the results of immunofluorescence analysis indicates a similar expression variation of vimentin after being treated by 3D-CS/Y@HMON. To further verify the hypothesis of EMT, Western blot was conducted to evaluate the associated molecular changes. As a well-known epithelial marker, the expression level of E-cadherin decreased after 24 h treatment with 3D-CS and 3D-CS/Y@HMON (Figure 6(d) and (e)), which related to the loss of the adherent epithelial phenotype. In contrast, the expression level of fibronectin and vimentin as mesenchymal markers appears an obvious up-regulation, thus confirming 3-D-CS/Y@HMON-induced EMT process in HPKs. 3D-CS-TEE and 3D-CS/Y@HMON-TEE were also verified had similar effects in vivo through analyzing tissue samples at day 7 after implantation. 3D-CS/Y@HMON-TEE treatment results in a significant decline of E-cadherin, as well as fibronectin, and an increase of vimentin, which further demonstrates EMT induced (Figure 6(f) and (g)). 38
Information exchange occurs among a variety of cells, including epithelial cells, in the process of wound healing, such as growth factor secretion by HPKs’ promoting migration and increasing the formation of new blood vessels in the wound, which could promote epidermis and dermis regeneration, accelerate wound epithelial healing, and improve the quality of wound healing. 10 TGF-β1 is an important cytokine that plays a key role in EMT. Previous studies found that TGF-β1 promoted EMT development in HPKs, whose morphology was transformed into fibroblast morphology. These HPKs expressed related surface markers and their migration was promoted after induction by TGF-β1. 39 On day 7 after 3-D-CS/Y@HMON-TEE transplantation, TGF-β1 expression in vivo was significantly higher than that in the 3-D-CS-TEE group in the wound tissue. However, TGF-β1 expression levels were decreased on day 14, while those in the 3-D-CS-TEE group were still rising (Figure 6(h)), which was possibly correlated with scar formation in the late repair stage. The differentiation of HPKs from the basal cell layer to the stratum corneum with the aid of the Y-27632-loaded nanoparticle platform requires further investigation.
The HPKs in 3-D-CS/Y@HMON-TEE were labeled with the cell membrane-labeling dye PKH26, which was detected on day 7 after implantation (Figure 6(i)), and imaged using a live animal in vivo spectroscopy system, indicating that the HPKs in the TEE indeed migrated. At this point, the intercellular movement of HPKs was no longer simply an extension to the center of the wound but migration from the inner TEE according to the reconstruction of the whole region. Compared with 3-D-CS-TEE, 3-D-CS/Y@HMON-TEE increased the migration and engrafting rates, but the relevant mechanisms guiding this approach were incorrect.
3-D-CS/Y@HMON-TEE accelerates epidermal histogenesis and tissue vascularization
During the wound repair process, establishing sufficient blood supply is the primary factor in skin regeneration. 40 Previous studies have shown that porous structural collagen with functionalized surfaces can provide a biomimetic 3-D microenvironment with the ability of drug molecules host to maintain drug release. 41 More crucially, the porous 3-D-CS structure may direct the migration and proliferation of fibroblasts and endothelial cells from the surrounding tissues, aiding the formation of granulation tissue and angiogenesis, and ultimately, wound healing. 42 We used immunofluorescence (Figure 7(a) and (b)) and qRT-PCR (Figure 7(e)) to evaluate the mRNA expression of VEGF and α-SMA in the wound tissues. The results revealed that on days 7 and 14, nascent tissues implanted with 3-D-CS/Y@HMON-TEE had considerably higher cytokine levels than those implanted with 3-D-CS-TEE (Figure 7(c)–(e)). VEGF, mainly synthetized and secreted by vascular endothelial cells and keratinocytes, is the most extensively studied angiogenic stimulus during wound healing. 43 α-SMA, a hallmark protein in myofibroblasts, is crucial for angiogenesis and wound healing. 44 Consistent with the in vitro experiments, 3-D-CS/Y@HMON-TEE significantly decreased the transcription of apoptosis-related proteins (Bax, Caspase-3, and Caspase-9) in nascent tissues, and the upregulation of the anti-apoptotic protein, Bcl-2, was statistically significant (Figure 7(f)). The above data strongly support that the TEE transplantation stably promotes wound healing in vivo, which was accelerated by HPKs migration and mesenchymal transformation. 45 3-D-CS provides a pro-angiogenic biological structure during wound regeneration, while the nanoparticle Y@HMON promotes epidermal histogenesis and tissue vascularization by ameliorating apoptosis. However, it was not clear if the anti-apoptotic effect was confined to specific cells.

3D-CS/Y@HMON-TEE in vivo reduces cells apoptosis to promote wound healing. (a–d) Immunofluorescence staining of α-SMA (green) and VEGF (red) at the wound site at different phases of wound healing on day 7 and 14. (e) The mRNA level of VEGF was measured via qt-PCR, 7 days after graft. (f) The mRNA expression level of Bax, Bcl-2, Caspase 3, and Caspase 9 detected by qt-PCR assay in groups. n = 3, ***p < 0.001.
Conclusion
In conclusion, we reported a TEE constructed with a nanoparticle (Y@HMON) and a 3-D collagen scaffold to reduce implanted cell apoptosis during construction and demonstrated its prominent performance in in vivo therapy. We discovered a new method in which nanoparticle platforms corrected the mitochondria-mediated apoptosis by alleviating the stress state in cells isolated from human skin. The nanoparticle platform significantly downregulated the ROS levels, turned off the mPTP to restore the mitochondrial transmembrane potential, and ultimately reduced the apoptosis rate. In addition, the sustained-release effect of the platform allows regulation throughout the process from TEE construction to the early stages of grafting, although the regulation in vivo requires further exploration. Unlike the conventional concept of organoid transformation, our work aims to construct a degradable scaffold based on advantageous biocompatibility with the property of sustained drug release to facilitate cell multiplication and accelerate wound healing through early vascularization. We believe that improving tissue regeneration and remodeling with the assistance of functional nanoparticles will arise great interest in complex wound repair, as it was a promising study to achieve the treatment of large-area skin defects.
Supplemental Material
sj-docx-1-tej-10.1177_20417314231163168 – Supplemental material for 3-D tissue-engineered epidermis against human primary keratinocytes apoptosis via relieving mitochondrial oxidative stress in wound healing
Supplemental material, sj-docx-1-tej-10.1177_20417314231163168 for 3-D tissue-engineered epidermis against human primary keratinocytes apoptosis via relieving mitochondrial oxidative stress in wound healing by Shan He, Han Wu, Junqun Huang, Qingyan Li, Zijie Huang, Huangding Wen and Zhiqing Li in Journal of Tissue Engineering
Footnotes
Abbreviations
3D: three dimensional; 3D-CS: three-dimensional collagen scaffold; α-SMA: a hallmark protein in myofibroblasts; BSA: bovine serum albumin; CCK-8: Cell Counting Kit-8 assay; CI: the collagen index; dSiO2: dense SiO2; ECM extracellular matrix; EMT: epithelial-mesenchymal transition; FITR: Fourier Transform infrared spectroscopy; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; H&E: hematoxylin and eosin; HMON: hollow mesoporous silica nanoparticles; HPK: human primary keratinocytes; JC-1: an ideal fluorescent probe widely used to detect mitochondrial transmembrane potential (ΔΨm ); LDH: lactate dehydrogenase; MOMP: mitochondrial outer membrane permeability; mPTP: mitochondrial permeability transition pore; mtROS: reactive oxygen species inside mitochondria; PBS: phosphate buffered saline; PVDF: polyvinylidene fluoride; qt-PCR: quantitative Real-time PCR; ROS: reactive oxygen species; SEM: scanning electron microscope; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; TEE: tissue-engineered epidermal; VEGF: vascular endothelial growth factor; XPS: X-ray photoelectron spectroscopy.
Author contributions
Zhiqing Li is responsible for conceptualization, project administration, funding acquisition. Shan He and Zhiqing Li led and designed the in vitro and in vivo experiment. Han Wu performed cell culture and collagen scaffold seeding. Shan He, Huangding Wen, and Junqun Huang performed animal experiments. Shan He, Zijie Huang, and Han Wu fabricated the collagen scaffolds. Han Wu and Qingyan Li analyzed the data and prepared the figures. Zhiqing Li and Shan He wrote the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors are grateful to the Science and Technology Planning Project of Guangdong Province (Grant Number 2017A020215042 to ZL). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research statement
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.