
Abstract
Herein, we report on the synthesis and characterization of enzymatically labile polyureas for use as a tissue-engineered ligament scaffold. Polyureas were selected due to their excellent tensile properties, fatigue resistance, and highly tunable nature. Incorporation of a collagenase-sensitive peptide into the backbone of the polyurea provided a means to confer cell-responsive degradation to the synthetic polymer. Chemical, morphological, and mechanical testing were used to confirm incorporation of the peptide and characterize polyurea films. Notably, the incorporation of the peptide resulted in an increase in modulus, elongation, and tensile strength. This was attributed to an increase in phase mixing and an increase in hydrogen bonding between the hard and soft segments. Candidate polyureas with varying levels of collagen-mimetic peptide (0%, 10%, 20%) were then subjected to degradation in collagenase media or buffer at 37°C over 4 weeks. Statistically significant decreases in strength and elongation were observed in polyureas with 20% peptide content after collagenase treatment, whereas specimens in phosphate-buffered saline showed no statistically significant difference. These observations confirmed that enzyme-specific degradation was conferred to the polyurea. Overall, these polyureas hold great promise as a material for ligament reconstruction due to the promising mechanical properties and potential for cell-mediated degradation.
Introduction
The anterior cruciate ligament (ACL) is the most frequently ruptured ligament, accounting for an estimated 200,000 injuries and approximately 100,000 reconstructive surgeries each year in the United States. Direct medical costs from surgical repair are estimated to be over 5 billion dollars. 1 Damage to the ACL can result in pain, loss of mobility, joint instability, and often leads to the development of degenerative joint diseases such as osteoarthritis. 2 Due to the poor vasculature and regenerative capability of ACL tissue, injuries often require surgical intervention.3–5 The current gold standard for ligament repair is an autograft that is typically harvested from the inner third of the patellar tendon or hamstring tendon. These grafts provide good mechanical strength and promote cell proliferation and differentiation.6–9 However, the autograft harvest procedure can lead to complications including donor site pain, tendonitis, muscle atrophy, and increased recovery time. 10 Alternatively, allografts typically utilize cadaveric ligaments or tendons that are processed and sterilized. 4 These grafts do not require harvesting of autogenous tissue, are more readily available than autografts, and are incorporated into the body much like autografts but at a slower rate. 8 However, allografts typically have reduced mechanical properties after processing and there is a risk of disease transmission.
Due to the limitations of biologic grafts, synthetic materials have been investigated for potential use in ACL reconstruction.11,12 These grafts are readily available and retention of mechanical properties allows for faster rehabilitation. 13 Synthetic ACL grafts are typically generated from non-degradable polymers including poly(tetrafluoroethylene) (PTFE), poly(ethylene terephthalate) (PET), and polypropylene.4,5,12,14–18 Although these grafts provide immediate stabilization of the joint, synthetic materials are unable to duplicate the mechanical behavior of the ACL and have a high fatigue failure rate.5,19 Additionally, the high linear stiffness of synthetic grafts causes a majority of the physiological load to be borne by the prosthesis, effectively stress shielding the surrounding tissue.5,20–22 Without proper mechanical cues to direct collagen alignment and tissue organization, the load-bearing capacity of the native tissue is reduced and the synthetic graft is limited to its inherent fatigue properties.
Musculoskeletal tissue engineering has received growing interest throughout orthopedic medicine as a promising alternative to biologic and synthetic grafts.16,22–24 ACL reconstruction utilizing a tissue-engineered ligament would eliminate donor site pain and morbidity, improve and accelerate rehabilitation, provide a limitless supply of graft tissue, eliminate the risk of disease transmission or unfavorable immunogenic responses, and increase the fatigue life of the graft.4,5 Polyesters such as poly(glycolic acid) and poly(lactic acid) as well as biological materials such as collagen and silk have been extensively studied for use in ACL reconstruction.2,23,25–29 Biodegradable materials have been shown to have highly tunable degradation and mechanical properties; however, these properties are typically linked such that one cannot be changed independent of the other. For example, reduced crystallinity has been used to increase degradation rate of polyesters and also results in reduced modulus.30,31 Furthermore, the requisite degradation rate that supports an appropriate load transfer in tissue is unknown and likely varies between patients. Improper selection of a degradation profile can lead to stress shielding if degradation is too slow or failure if degradation is too rapid. In contrast, enzymatic degradation of the polymer would circumvent this issue by conferring cell-responsive degradation to the graft and insert it into the native remodeling process.
In order to integrate with native ligament remodeling and maintain mechanical functionality, we have developed a polyurea elastomer with collagenase-sensitive moieties. Polyurethane and polyurea elastomers have been used in a variety of biomedical applications over the past 40 years and have received growing interest in tissue engineering applications.32–35 Polyurethane and polyurea chemistry dictates the physical, biological, and mechanical properties of these polymers and can be tailored to provide a variety of materials, such as soft elastomers, rigid thermosets, and foams.33,36–38 Due to the exceptional tunability of polyurethanes and polyureas, segmental modification of these polymers can be used to generate a library of polymers with broad structural diversity and a myriad of performance properties to better probe specific tissue–biomaterial interactions. Biodegradable formulations typically incorporate hydrolytically labile hard and soft segments or enzymatically labile hard segments.39–51 However, degradation of hard segments is highly dependent on crystallinity, which is a well-established barrier to degradation. Biodegradable soft segments can potentially decouple the effects of polyurethane structure on degradation rate and mechanical properties. Several polyurethanes composed of hydrolytically labile soft segments such as polyesters have demonstrated biocompatibility and excellent mechanical properties, as well as promoted tissue remodeling.52,53 However, the semi-crystalline nature of some of these soft segments can impact the performance properties of these materials.43,44
For this study, a polyurea elastomer was synthesized that incorporates a collagen-derived peptide into the soft segment to confer cell-responsive degradation to the polymer. A triblock soft segment structure was designed with poly(tetramethylene glycol) (PTMG) on the ends and a short polyethylene glycol (PEG) linker unit in the center that served as an analog for the peptide in structure–property investigations. For biodegradable formulations, a percentage of this PEG linker is substituted for the peptide which is approximately equivalent in molecular weight and similar in hydrophilicity. The objective of this design was to reduce the change to the polyurea structure and decouple degradation rate from the mechanical properties. Polyureas with varying levels of peptide were characterized to determine the effect of the peptide on morphology, mechanical properties, and rate of enzymatic degradation. In addition to the generation of tough elastomers with cell-responsive degradation, the structure–property models presented here are expected to aid in the elucidation of load transfer mechanisms critical to ligament regeneration.
Materials
Polyether diamines were acquired from Huntsman Corporation (The Woodlands, TX, USA). PEG diamine (RE-900, Mn = 900 Da) and PTMG diamine (XTJ-548, Mn = 1700) were used. Polymers were azeotropically dried by dissolving in toluene at a concentration of 100 mg/mL followed by solvent removal using rotary evaporation at 100°C. Hexane diisocyanate (HDI), ethylene diamine (EDA), 1,2-diaminopropane (2DAP), 1,3-diaminopropane (3DAP), and lithium bromide (LiBr) were obtained from Sigma–Aldrich and used as received. Anhydrous dimethylformamide (DMF) was obtained from Sigma–Aldrich and stored over molecular sieves. Phosphate-buffered saline (PBS) and penicillin–streptomycin (Pen–Strep) solution (10,000 meq/mL, 10 mg/mL) were obtained from Sigma–Aldrich and used as received. Collagenase IV was obtained from West Labs in New Haven, CT, and used as received. The collagen-mimetic peptide diamine (GPQGIWGQGK–CONH2) was obtained from Celtek Peptides in Nashville, TN, and used as received. The peptide was produced with a free amine on the N-terminus, an amide blocked C-terminus, and a second free amine located on the lysine side chain.
Methods
Polyureas were created in a one-pot synthesis with serial addition of reagents. Reactions were performed in a reaction vessel under a dry nitrogen blanket with vigorous stirring. Polyether diamines were first dissolved in DMF at a concentration of 25 mg/mL. The polymer solution was then added to a 50 mg/mL solution of HDI in DMF, forming the diisocyanate-functionalized prepolymer. The linking peptide diamine was then dissolved in DMF at a concentration of 25 mg/mL and added to the prepolymer diisocyanate solution in biodegradable formulations. The linking PEG diamine also dissolved in DMF at a concentration of 25 mg/mL and added to the prepolymer diisocyanate solution in all formulations. A 10-mg/mL solution of the chain extender in DMF was then added drop-wise to build molecular weight (Figure 1). Polyureas were formulated with 15% hard segment (HS) content, composed of a 1:1 mixture of 2DAP. HS content was calculated from the HDI and diamines located in the HS blocks; this did not include HDI linking soft segment blocks.

Synthetic design of enzymatically degradable polyureas.
All spectra were normalized to the 1591 cm−1 peak corresponding to the C=C bond stretch of the aromatic ring in the hard segment. Fourier transform infrared (FTIR) spectroscopic analysis was performed on a Bruker TENSOR 27 spectrometer (Billerica, MA, USA) equipped with a 45° germanium crystal to confirm reaction completion, extent of hydrogen bonding, and incorporation of the peptide. Reacted polymer solutions were cast onto glass Petri dishes at 50°C under vacuum to a thickness of approximately 0.2 mm. Tensile testing was performed with an Instron 3345 Single Column Universal Testing System equipped with a 1-kN load cell and 250 N pneumatic grips, tested at 100% strain/min. Dynamic mechanical analysis (DMA) was performed using a TA Instruments RSA3 instrument to investigate relative phase separation. Temperature sweep tests were performed from −90°C to 150°C with a heating rate of 5°C/min while under a 0.1% cyclic strain at 1-Hz frequency.
Polymer films were subjected to enzymatic degradation for up to 4 weeks and then examined for chemical, mechanical, and visual changes (n = 3). Specimens were submerged in 37°C PBS solution with or without collagenase (0 mg/mL, 10 mg/mL). All solutions contained 1% Pen–Strep solution to prevent microbial contamination. Solutions were changed and specimens were weighed weekly. Uniaxial tensile testing of samples was performed after 2 and 4 weeks and was compared to control specimens. Scanning electron microscopy (SEM) was used to inspect polyurea films for surface damage.
Results and discussion
First, successful incorporation of the peptide into the polyurea was confirmed with FTIR spectroscopy. Spectra of polymer films contained peaks correlating to hydrogen-bonded N–H stretching (3300 cm−1), the methyl backbone of PTMG and PEG (2900 cm−1), amide from the peptide (1660 cm−1), urea (1650 cm−1), hydrogen-bonded urea (1617 cm−1), and the ether backbone of PTMG and PEG (1080 cm−1; Figure 2). Although the peptide peak is convoluted by the urea peak, a trend was observed that indicated successful peptide incorporation. A shoulder at 1660 cm−1 assigned to the amide of the peptide increased with increasing peptide content. Intensities at 1660 cm−1 were observed to increase by 32% and 48% for polyureas with 10% and 20% peptide content, respectively. The presence of hydrogen-bonded urea also suggests that the peptide did not disrupt the two-phase morphology.

FTIR spectra of PTMG polyureas with 15% hard segment content and 0%, 10%, and 20% peptide content.
Effect of peptide on morphology and mechanical properties
To assess phase morphology, DMA plots of polyureas with varying peptide content were compared (Figure 3). A broadening of the glass transition (Tg) with increasing peptide content was observed and was attributed to an increase in phase mixing. Given that the peptide contains several amide groups that function as hydrogen bond donors (N–H) and acceptors (C=O), it was hypothesized that the peptide provided an increased opportunity for hydrogen bonding between the hard and soft segments.

Dynamic mechanical analysis of PTMG polyureas with 15% hard segment content and 0%, 10%, and 20% peptide content.
Stress–strain plots were analyzed to investigate the impact of the peptide and its observed differences in morphology on tensile properties (Figure 4). PTMG polyureas all displayed a yield point followed by drawing. A table of mechanical properties is provided (Table 1). Overall, the tensile properties of polyureas with varying peptide content showed increased tensile strength and elongation with increasing peptide content. The increase in phase mixing evident in the DMA with increasing peptide content was hypothesized to have been a result of the peptide hydrogen bonding with the hard segment. This increased phase mixing likely permitted continued chain organization and strain-induced crystallization, resulting in the observed increase in the ultimate tensile strength.

Stress–strain behavior of polyureas with 15% hard segment content and 0%, 10%, and 20% peptide content.
Tensile properties of untreated polyureas.

Enzymatic degradation of biodegradable polyureas
Polymer films were subjected to degradation in collagenase solutions over a period of 4 weeks to determine the enzyme-specific degradation of the peptide-based polyureas. FTIR spectroscopic analysis was used to evaluate peptide degradation. A detailed view of the carbonyl region of the polyureas is displayed in Figure 5. A decrease in peak height at 1660 cm−1 assigned to the peptide was observed after 4 weeks of degradation in polyureas with 20% peptide content; however, no corollary decrease was discernible in polyureas with 10% peptide content. Subtracting the 0% peptide as a baseline, polyureas with 20% peptide content experienced a 40% reduction in the peak at 1660 cm−1 indicating possible chain scission of the peptide and extraction of low-molecular-weight species. No spectral changes were detected in polyureas with 10% or 20% peptide when subject to degradation in PBS without collagenase. This indicates that the observed changes in the peptide-based polyurea were specific to enzymatic chain scission.

FTIR analysis of PTMG-based polyureas containing 15% hard segment content and 0%, 10%, and 20% peptide content after 4 weeks of degradation in PBS/collagenase solution (carbonyl region).
Uniaxial tensile testing of specimens subjected to 4 weeks of degradation was analyzed to investigate the impact of degradation on the tensile properties. Table 2 provides a summary of the tensile properties values obtained from the degradation study. The values are given for the untreated control as well as 2 and 4 weeks in PBS and collagenase solutions. For polyureas with 0% peptide content, there was no significant change in modulus, elongation, or strength when subject to degradation in PBS or collagenase as compared to untreated samples. For polyureas with 10% peptide content, a small decrease in tensile strength and elongation was observed at 4 weeks in collagenase, but this difference was not statistically significant. For polyureas with 20% peptide content, a statistically significant decrease in tensile strength and elongation was observed at both 2 and 4 weeks. It is important to note that this difference was only present in films subjected to degradation in collagenase, not PBS alone. No statistically significant changes were observed in any samples subject to degradation in PBS at 2 or 4 weeks as compared to untreated samples.
Tensile properties of polyureas after 2 and 4 weeks of degradation in PBS or collagenase solution.
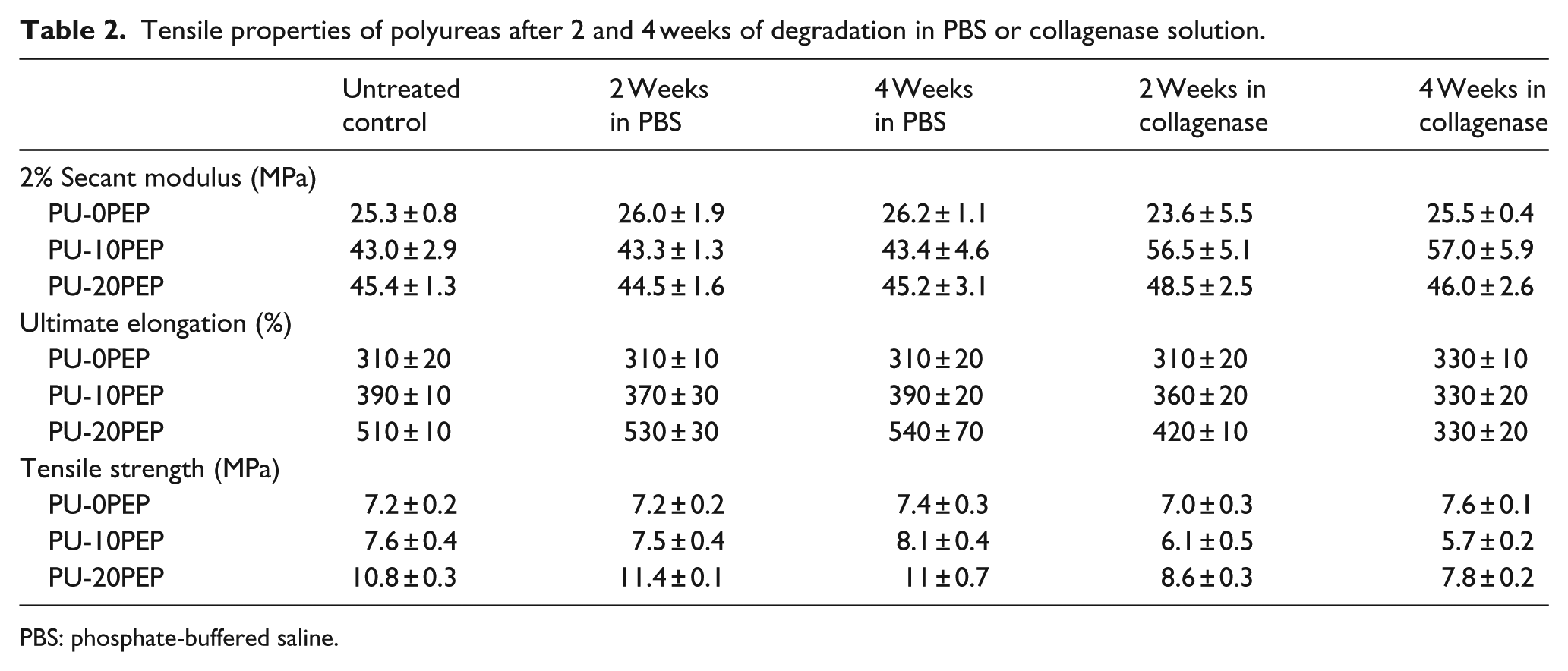
PBS: phosphate-buffered saline.
The percent changes in strength, elongation, and modulus are displayed in Figures 6–8, respectively. These results correlate well with the infrared findings that indicate cleavage of the peptide. Peptide degradation results in lower molecular weight, correlating to lower ultimate tensile strength, as discussed previously. A reduction in ultimate elongation is explicated by peptide degradation as well. Overall, the reduction in the mechanical properties was attributed to peptide cleavage shown in FTIR analysis.

Percent change in ultimate tensile strength of specimens after 2 and 4 weeks of degradation in PBS/collagenase solution.

Percent change in ultimate elongation of specimens after 2 and 4 weeks of degradation in PBS/collagenase solution.
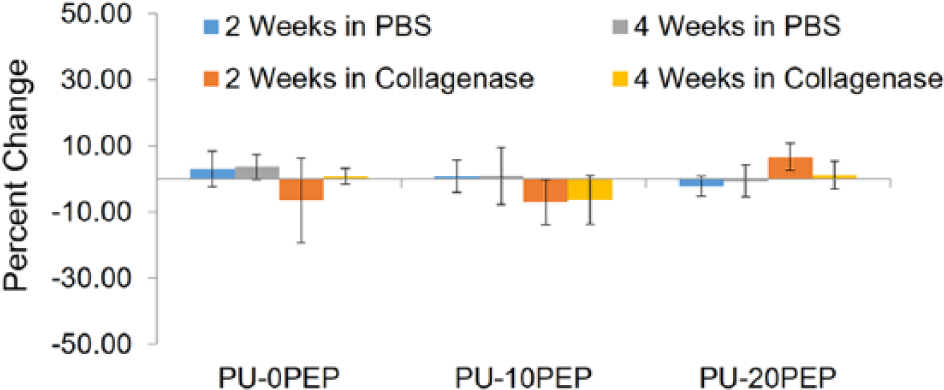
Percent change in 2% secant modulus of specimens after 2 and 4 weeks of degradation in PBS/collagenase solution.
SEM was used to analyze the surface of the degraded and control polymer films (Figure 9). Polyurea specimens with 0% peptide were found to have a very smooth topography, with no visible difference between the untreated and samples aged in collagenase for 4 weeks. The 20% peptide films were observed to have a smooth surface before degradation, but show a rough, patterned surface after being subject to degradation in collagenase solution for 4 weeks. The surface roughness is likely due to surface degradation. Enzymatic degradation is expected to be localized to the surface because enzymes are typically too large to diffuse into the polymer and cause bulk degradation.44,54,55 The observed surface degradation is concordant with the changes in the chemical and tensile properties. The bond cleavage and reduction in strength supports a possible reduction in molecular weight; however, due to the extent of hydrogen bonding, films could not be dissolved for molecular weight analysis with gel permeation chromatography (GPC). Despite the evidence of surface degradation, no significant mass loss was observed at these time points.

SEM images of polyurea film surface damage after 4 weeks of degradation in PBS/collagenase solution.
Conclusion
In summary, we have characterized the physical properties of a novel biodegradable polyurea and subjected it to enzymatic degradation. The results of this study indicate successful synthesis of polyureas with enzymatic degradation. Further elucidation of key structure–property relationships is necessary to determine the effect of composition on microphase-separated morphology. Ultimately, understanding such relationships will be critical for the development of an improved tissue-engineered ligament for ACL reconstruction. These polyureas combine the strength and tunability of synthetic elastomers with the cell-responsive degradation of native collagen. The ultimate tensile strength of these materials (7–10 MPa) is within an order of magnitude of the human ACL, ~38 ± 9 MPa. 18 Although the ligament graft does not match native tissue, it is expected to have sufficient strength to restore initial function and withstand physiological loading until the graft is remodeled to match native tissue properties. This hybrid design integrates the graft into native ligament remodeling and may facilitate load transfer from the biodegradable scaffold to neotissue at a rate that promotes proper tissue orientation and function while maintaining construct integrity. The addition of cell-responsive degradation to one of the most versatile classes of biomaterials makes these hybrid grafts promising. In addition to the development of an improved biomaterial for ACL reconstruction, synthetic strategies used to generate a library of cell-responsive, biodegradable polyureas can be utilized for a variety of other biomedical applications as well.
Similar to the current approach, enzyme-labile polyureas can be developed to create new structure–property models for bone or cardiovascular tissue engineering. A polymeric system that combines the tunability of segmented block copolymers with system-responsive degradation can also be used to achieve effective drug delivery.56,57 Based on the versatility of the synthetic routes described above, enzyme-labile peptide sequences can be replaced with other sequences to produce an assortment of biomimetic materials. Overall, the synthesis of a library of cell-responsive, biodegradable polyureas will assist in the development of a tissue-engineered ligament, as well as provide additional tools to advance biomaterial design.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.