
Abstract
Functional tissue engineering for bone augmentation requires the appropriate
combination of biomaterials, mesenchymal stem cells, and specific
differentiation factors. Therefore, we investigated the morphology, attachment,
viability, and proliferation of human dental pulp stem cells cultured in
xeno-free conditions in human serum medium seeded on β-tricalcium
phosphate/poly(
Keywords
Introduction
Considerable bone loss due to trauma, periodontitis, resorption of edentulous maxillary ridge, and delayed healing of the extraction sockets compromises placement of dental implants. 1,2 It is known that sufficient bone augmentation is a prerequisite for placement of dental implants to achieve functionality and long-term treatment outcomes. 3 Traditionally, for repairing such defects, autogenous or allogenic bone grafts are harvested and implanted into the affected areas. 4 However, due to the clinical drawbacks including donor site morbidity and risk of transmitted diseases, there is a need for alternative solutions such as tissue-engineered bone grafts. 5 Tissue engineering is an interdisciplinary approach to repair damaged tissue. This approach regenerates the tissue through the use of biodegradable implants combined with in vitro cultured stem cells and differentiation factors. 6,7 Mesenchymal stem cells (MSCs) are a unique and easily isolated source of cells for bone tissue engineering. Various sources for MSCs have been reported, including the bone marrow (BM), cord blood (CD), and adipose tissue (AD), and their potential in bone regeneration has been studied extensively in vitro 8 –10 and in clinical applications. 2,6
Recently, there has been emerging inclination toward studying another source of MSCs obtained from teeth such as dental pulp tissue for its ability to regenerate bone. 11 Dental pulp stem cells (DPSCs) can be isolated from the dental pulp tissue, which is obtained from the impacted third molar teeth with ease and minimal tissue site morbidity. The dental pulp tissue is a loose connective tissue that provides nutritional and sensory properties to dentin and has reparative capacity to form tertiary dentin; therefore, it is believed to possess stem/progenitor cells. 12 Several research reports have shown clonogenic ability, rapid proliferative rate, and multiple differentiation ability of DPSCs including two studies from our group. 13 –15 Most commonly, osteogenic differentiation potential of DPSCs has been induced by dexamethasone (DEX), which is a synthetic glucocorticosteroid. 16 Another osteogenic inducer, 1α,25-dihydroxyvitamin D3 (vitamin D3 (VD)), in combination with β-glycerophosphate and ascorbic acid has been reported as more potent than DEX for DPSC differentiation in our previous study 14 and in another study for AD-MSCs differentiation. 17 Apart from the osteogenic inducers, high fetal bovine serum (FBS) concentration has been shown to induce osteogenesis, wherein a subpopulation of DPSCs were capable of forming woven bone in vitro. 18 Intriguingly, in a clinical study, it was shown that DPSCs isolated in FBS containing medium seeded onto collagen sponge scaffolds and implanted in the tooth extraction socket hastened the process of bone regeneration. 19 Nevertheless, there are concerns regarding the use of FBS and other animal-derived supplements in culturing cells for clinical therapy due to risk of transmitting zoonoses in humans. 20,21 Thus, alternative serum supplementation is needed; the possible choice would be autologous or pooled allogenic human serum (HS), which can be tested for human pathogens before use. 22,23
A range of biomaterials have been studied to investigate the MSC attachment, growth,
and osteogenic differentiation.
24,25
Collagen sponge in combination
with DPSCs has been successfully used to regenerate bone in tooth extraction
sockets.
19
However, it has been shown that collagen sponge as a scaffold
has poor mechanical strength and high dimensional changes.
26
On the other hand,
osteoconductive, bioresorbable, bioceramic material β-tricalcium phosphate
(β-TCP) has been used for bone tissue engineering applications in clinics
for years but this material has poor mechanical strength.
27,28
To overcome the mechanical
strength drawback of bioceramics, combination of β-TCP and
poly(
Thus, the aim of this study was to assess DPSC adhesion, proliferation, and osteogenic differentiation within β-TCP/P(LLA/CL) three-dimensional (3D) scaffolds in clinically applicable conditions by using HS as a medium supplement to replace FBS. Furthermore, the osteogenic differentiation of the DPSCs within the β-TCP/P(LLA/CL) biomaterial scaffold induced by VD or DEX was compared under xeno-free conditions.
Material and methods
Cells isolation and culture
Human-impacted third molars were obtained with informed consent from Finnish
Student Health Services, Tampere, Finland. The collection of stem cells from
tooth samples was approved by the Ethics Committee of the Pirkanmaa Hospital
District, Tampere, Finland (R06009). Human dental pulp explants were obtained
from partially impacted third molar teeth (Figure 1(a)) of three patients aged
21–26 years (23 ± 2.5 years). The pulp tissue explants (Figure 1(b)) were
transported to the laboratory in Dulbecco’s phosphate-buffered saline
(DPBS; BioWhittaker; Lonza, Verviers, Belgium) containing 2%
antibiotics/antimycotics (a/a; 100 U/mL penicillin, 0.1 mg/mL streptomycin, and
0.25 µg/mL amphotericin B; Life Technologies, Paisley, Scotland). The
dental pulp tissue fragments were minced by using scalpels and were then
digested in collagenase type I (3 mg/mL; Invitrogen) and dispase (4 mg/mL;
Invitrogen, Paisley, Scotland, UK) for 1 h at 37°C. Once digestion was
completed, the obtained cell pellet was suspended in 500 µL of DPBS and
was passed through a 100-µm cell strainer (Falcon; BD Labware, Franklin
lakes, NJ, USA). The isolated DPSCs were cultured and expanded in
Dulbecco’s modified Eagle’s medium (DMEM)/F-12, 1:1 mixture
(Gibco Life Technologies, Paisley, UK) supplemented with 1%

(a) Representative pictures of extracted human-impacted third molar tooth to obtain (b) dental pulp tissue. (c) After 14 days, the cell morphology appeared spindle shaped as observed under phase contrast microscope, scale bar = 100 μm.
After initial passaging, the concentration of HS was reduced to 15% in the culture medium. Cell culture plates and flasks were monitored daily for cell growth, with medium changes taking place three times per week. All assays were performed using cells between passages 2 and 4, and all experiments were repeated using DPSCs derived from three different donors (n = 3).
Flow cytometric surface marker expression analysis
DPSCs cultured in HS-M were analyzed for cell surface antigen expression by flow cytometry (fluorescence-activated cell sorting (FACS); FACSAria®; BD Biosciences, Erembodegem, Belgium). Monoclonal antibodies (MAb) against CD90-Allophycocyanin (APC), CD105-phycoerythrin (PE) (R&D Systems Inc., Minneapolis, MN, USA), CD31-fluorescein isothiocyanate (FITC) (Immunotools GmbH, Friesoythe, Germany), and CD45-FITC (Miltenyi Biotec, Bergisch Gladbach, Germany) were used. Antibodies were added to 100,000 cells/sample and then incubated for 30 min at 4°C in the dark. After incubation, cells were washed and then analyzed by flow cytometry.
Preparation and evaluation of the biomaterial by scanning electron microscope
β-TCP/P(LLA/CL) (ChronOS™) was provided by Synthes (Oberdorf, Switzerland); the material is accepted for clinical use as a bone graft substitute. The biomaterial was supplied in sterile strip form with a size of 2.5 cm × 5 cm (Figure 2(a)): two 3-mm-thick strips and three 6-mm-thick strips. For the experiments, the strips were cut into 1 cm × 0.8 cm pieces (Figure 2(a)) with scalpels in sterile conditions under the laminar flow hood.
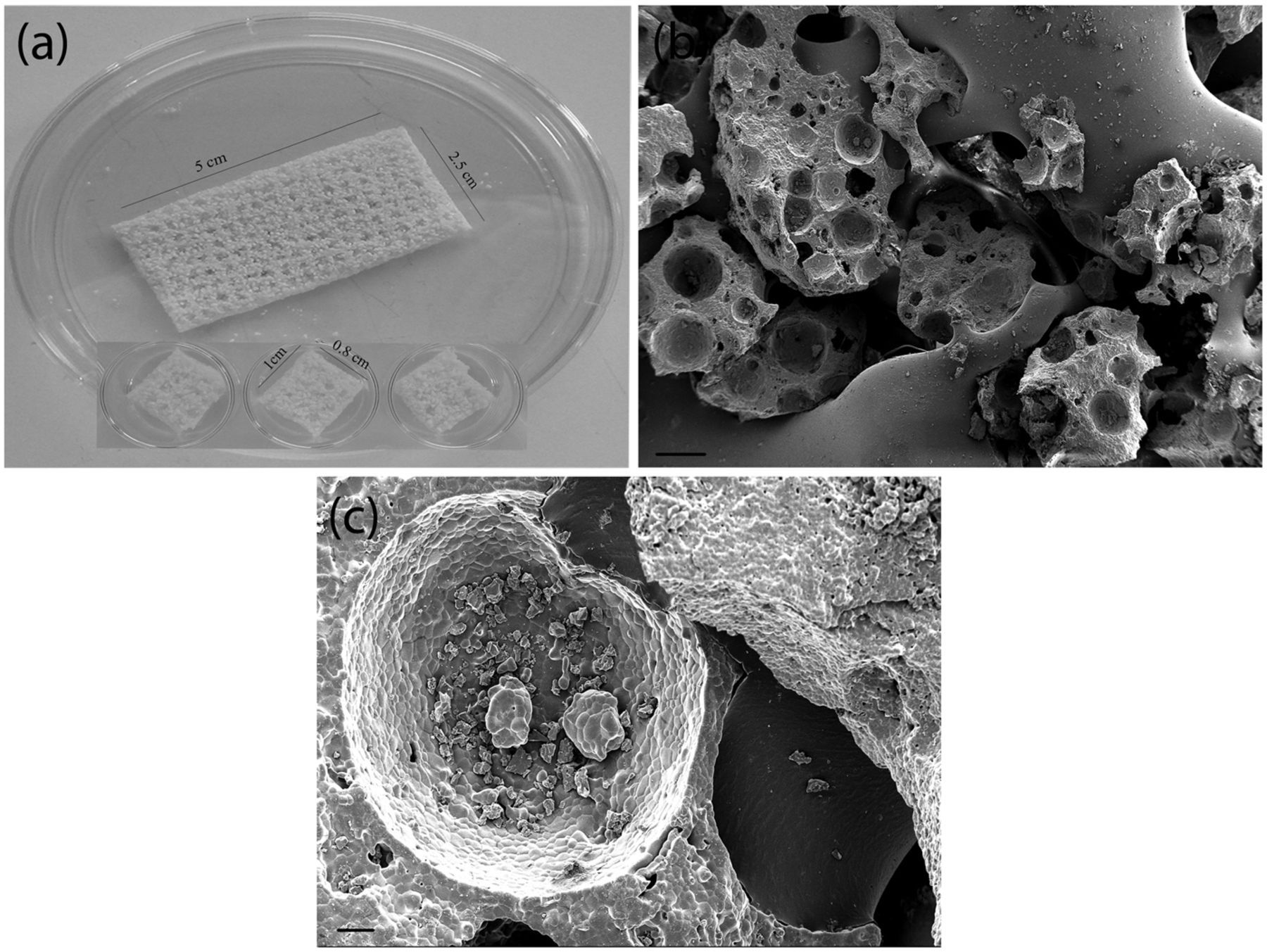
(a) Representative pictures of the β-TCP/P(LLA/CL) (ChronOS) biomaterial strip and the biomaterial pieces cut into 1 cm × 0.8 cm dimension for in vitro analysis. (b) Scanning electron microscope analysis showing β-TCP particles embedded in P(LLA/CL) and (c) the fine structure of β-TCP particle, scale bar = 20 and 100 μm.
Subsequently, for scanning electron microscope (SEM) analysis, the biomaterial samples were rinsed with DPBS and dehydrated through a series of ascending concentration of ethanol (30%, 50%, 70%, 90%, and 100%). The samples were then incubated in hexamethyldisilazane (HMDS) for 10 min and dried overnight in a dessicator. The dried biomaterial scaffolds were cut into half and mounted on a double-sided carbon tape. A platinum coating was sputtered on the samples before SEM observation.
Cell seeding and treatment conditions
The biomaterial scaffold pieces were transferred into 24-well plates, washed with
DPBS, and incubated in HS-M at 37°C in 5% CO2 for assisting
in attachment of cells before cell seeding. After 48 h of incubation, the
scaffolds were seeded with 300 cells/mm3, and 150 µL of cell
suspension was added onto each biomaterial scaffold. Cells were allowed to
attach within the porous scaffold for 2 h before adding 500 µL of the
culture or differentiation medium. Osteogenic medium (OM), containing HS and
either of the hormones dexamethasone (OM-DEX; 10 nM) or
1α,25-dihydroxyvitamin D3 (OM-VD; 100 nM) in addition to 50
µM
Cell attachment and viability
Cell attachment and viability of DPSCs in biomaterial scaffolds cultured in HS-M were evaluated at days 1, 7, and 14 using live/dead-cell staining kit (Molecular Probes, Eugene, OR, USA) according to the manufacturer’s protocol. In brief, cell-seeded scaffolds were incubated in DPBS-based dye solution, containing 0.5 µM of calcein AM (green fluorescence; Molecular Probes) (4 mmol/L) and 0.5 µM of ethidium homodimer-1 (EthD-1; red fluorescence; Molecular Probes) (2 mmol/L) for 45 min at room temperature (RT). The dye solution was replaced by fresh DPBS solution. The viable cells (green fluorescence) and necrotic cells (red fluorescence) were examined using a fluorescence microscope.
CyQUANT® cell proliferation assay
CyQUANT® Cell Proliferation Assay Kit (CyQUANT; Molecular Probes, Invitrogen) was used according to the manufacturer’s protocols to assess the cell numbers at 1, 7, and 14 days. Cell-seeded scaffolds were cultured in OM-DEX, OM-VD, and HS-M. In brief, 500 µL of 0.1% Triton X-100 (Sigma) was pipetted through the cell-seeded scaffolds and the lysed cell suspensions were frozen until analysis. The CyQUANT cell proliferation assay is based on the green fluorescence dye, CyQUANT GR dye, which intensifies when it binds to the nucleic acid of DNA. The fluorescence, which is directly proportional to the number of cells in the sample, was measured at 480/530 nm using a microplate reader (Victor 1420 Multilabel Counter; Wallac, Turku, Finland).
Alkaline phosphatase staining
In vitro osteogenic differentiation capacity of the DPSCs induced by OM-DEX and OM-VD was determined at day 14 by alkaline phosphatase (ALP) staining. The control cell-seeded scaffolds were cultured in HS-M. Cell-seeded scaffolds were stained by using a leukocyte ALP kit according to Sigma procedure 86 (cat. no. 86R-1KT). In brief, cell-seeded scaffolds were fixed with 4% paraformaldehyde (PFA) solution for 2 min. ALP staining solution was added to the scaffolds following the fixation. After 15 min of incubation in the dark, the ALP staining solution was removed and the scaffolds were washed to remove excess stain. Thereafter, digital images were taken of the ALP stained and unstained scaffolds.
Immunostaining
After 14 days of inducing osteogenic differentiation in the cell-seeded scaffolds using OM-DEX and OM-VD, they were immunostained with primary antibody antihuman osteocalcin (OCN) (AbD Serotec, Immunodiagnostics Oy, Finland). The cell-seeded scaffolds cultured in HS-M were used as controls. In brief, the cell-seeded scaffolds were fixed with 4% PFA for 10 min and then blocked against nonspecific antigen binding with 10% normal donkey serum (NDS), 0.1% Triton X-100, and 1% bovine serum albumin (BSA) in DPBS. After 45 min of blocking, the cell-seeded scaffolds were washed with 1% NDS, 0.1% Triton X-100, and 1% BSA in DPBS (washing solution). The primary antibody antihuman OCN was diluted to 1 : 50 in the washing solution. The cells were incubated overnight at +4°C with the primary antibody. The next day, the cells were washed with 1% BSA in DPBS and incubated for 1 h at RT. Thereafter, cells were incubated for 1 h in Alexa Fluor-488 (1 : 1000; Invitrogen) conjugated anti-mouse secondary antibody, diluted in 1% BSA in DPBS. Then, cells were sequentially washed with PBS and phosphate buffer and mounted with Vectashield (4′,6-diamidino-2-phenylindole (DAPI); Vector Laboratories, Peterborough, UK). For negative controls, primary antibody was omitted. Stained DPSCs within the scaffolds were imaged using a microscope equipped with a fluorescence unit and camera.
Quantitative real-time polymerase chain reaction
The cell culture conditions were same as described for ALP staining. The control samples were maintained in the HS-M. The total RNA was extracted at 7 and 14 days using Eurozol (Euroclone S.p.A, Pero, Italy). First-strand complementary DNA (cDNA) syntheses were performed by a High Capacity cDNA Archive Kit (Applied Biosystems, Warrington, UK). Quantitative real-time polymerase chain reaction (qRT-PCR) was conducted using primers for human acidic ribosomal phosphoprotein (RPLP0), OCN, osteopontin (OPN), and ALP (Table 1). To exclude signals from contaminating DNA, the forward and reverse sequences of each primer were designed on different exons. The Power SYBR Green PCR Master Mix (Applied Biosystems) was used for quantitative PCR reactions according to the manufacturer’s instructions. The reactions were performed with ABI Prism 7300 Sequence Detection System (Applied Biosystems) at 95°C/10 min, and then 45 cycles at 95°C/15 s and 60°C/60 s were performed. The Ct values for OCN, OPN, and ALP were normalized to that of the housekeeping gene RPLP0, as described elsewhere. 34
Primer sequences for quantitative RT-PCR

RT-PCR: real-time polymerase chain reaction.
Statistical analysis
The statistical analyses of the results were performed with GraphPad Prism 5.01. The data are presented as mean ± standard deviation (SD) for all quantitative assays, and experiments were carried out in triplicate for cells derived from three donor samples. All statistical analyses were performed at the significance level p < 0.05 using one-way analysis of variance (ANOVA) with Bonferroni post hoc test for multiple comparisons.
Results
Cell isolation and morphology
From day 1 to day 9, DPSCs cultured in HS-M proliferated in colonies and spherical clusters. Morphologically, cells mostly appeared spindle shaped and comprised a homogenous cell population when viewed under the phase contrast microscope (Figure 1(c)). After first passage, the cells did not proliferate in clusters; rather, cells were more spread out and proliferated uniformly.
Flow cytometric surface marker expression analysis for human DPSCs
Prior to seeding the cells into the biomaterial scaffolds, flow cytometric assay was done to define the mesenchymal surface marker expression of the cells. The DPSCs were strongly positive for MSC markers CD90 and CD105 and were negative for hematopoietic lineage markers CD31 and CD45 (Figure 3).

Surface marker expression of dental pulp stem cells cultured in human serum medium as analyzed by flow cytometry. Histograms demonstrating the relative cell count (y-axis) and fluorescence intensity (x-axis), with unstained control cells (empty histogram) and cells stained with antibodies against the surface proteins CD90, CD105, CD31, and CD45 (filled histogram).
Cell attachment and viability
The cell-seeded scaffolds cultured in HS-M were analyzed for cell viability. The results revealed that DPSCs were viable, attached, and migrated into the pores of the biomaterial scaffold following day 1, day 7, and day 14 time course, with very few dead cells. Visually, upon viewing microscopically, more cells were observed at day 7 and day 14 in comparison to day 1 (Figure 4).

Representative images of viable and dead DPSCs seeded on β-TCP/P(LLA/CL) 3D scaffolds at (a) day 1, (b) day 7, and (c) day 14 (scale bar = 500 µm). Viable cells stained green and dead cells stained red after calcein AM/EthD-1 staining. The cells were seen to remain viable after day 7 and day 14 of culture in β-TCP/P(LLA/CL) as observed under the fluorescence microscope (n = 3) (online version in color).
Cell proliferation
The cell-seeded scaffolds cultured in HS-M, OM-DEX, and OM-VD were assessed for increase in cell numbers from day 1 and day 7 to day 14. There was significant increase in cell numbers at day 7 (p < 0.01) and day 14 (p < 0.001) time points, when cells cultured in OM-VD were compared to the control HS-M at days 7 and 14 (Figure 5). Though there was no significant increase in cell numbers when cells were cultured in OM-DEX, the cells numbers increased nonsignificantly. Moreover, there was significant increase (p < 0.001) in cell numbers cultured in OM-VD when compared to OM-DEX at day 14.
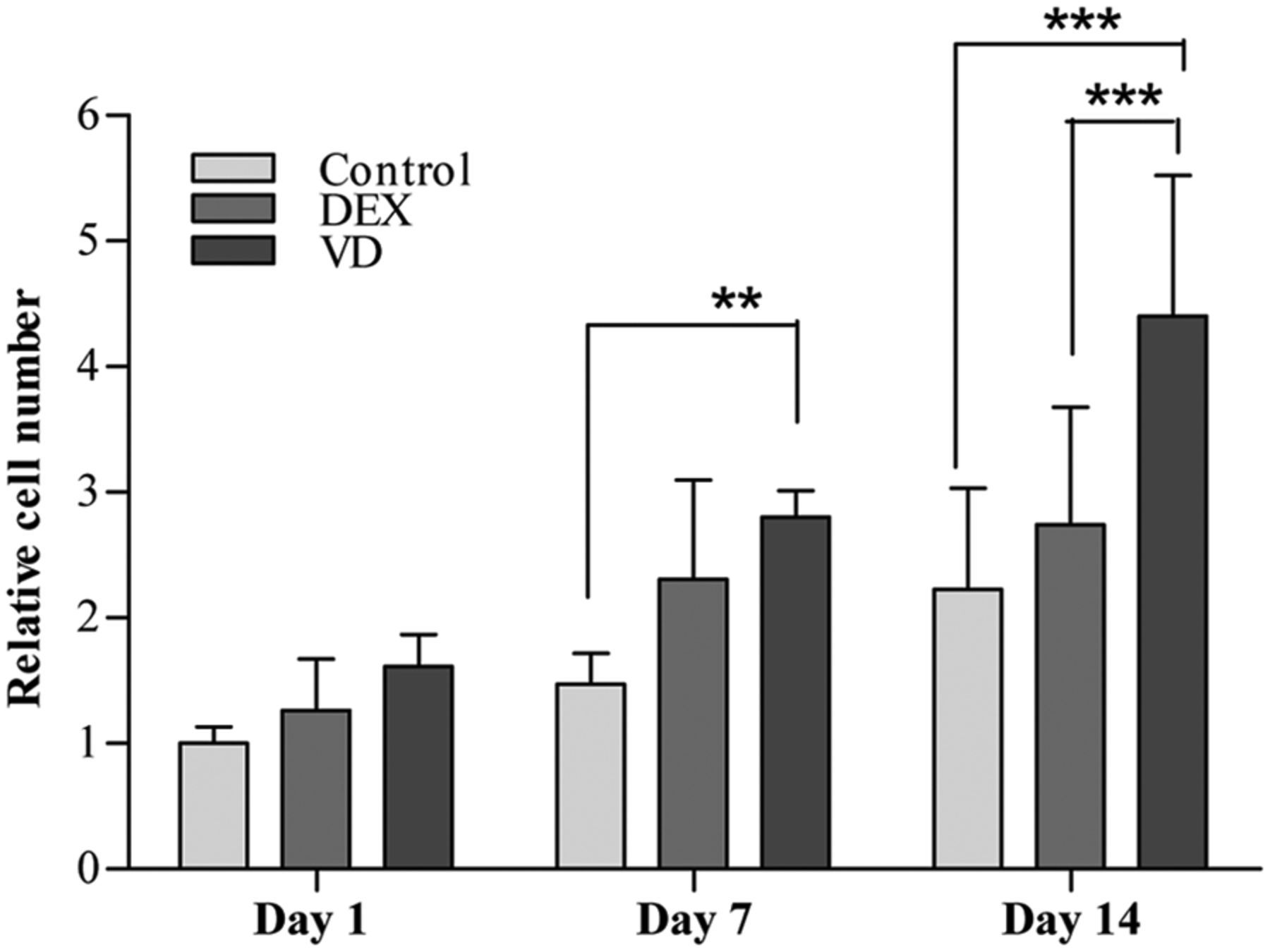
Cell growth characteristics of DPSCs seeded on β-TCP/P(LLA/CL) 3D scaffolds at days 1, 7, and 14 assessed by measuring the DNA content.
ALP staining and messenger RNA expression of ALP
ALP is an early marker that indicates differentiation of cells toward osteogenic lineages. 35 The ALP activity of cells cultured in HS-M, OM-DEX, and OM-VD is depicted in (Figure 6(a) to (c)). Interestingly, ALP activity expressed by cells cultured in OM-VD was stronger than the cells cultured in OM-DEX. The control cells were cultured in HS-M and no ALP staining could be detected. Subsequently, similar findings were observed when ALP expression was analyzed at mRNA level. ALP expression was significantly induced at day 7 (p < 0.05) and day 14 (p < 0.05) when the DPSCs treated with OM-VD were compared with control sample. However, no significant differences were observed when cells treated with OM-DEX and OM-VD were compared, as shown in (Figure 6(d)).
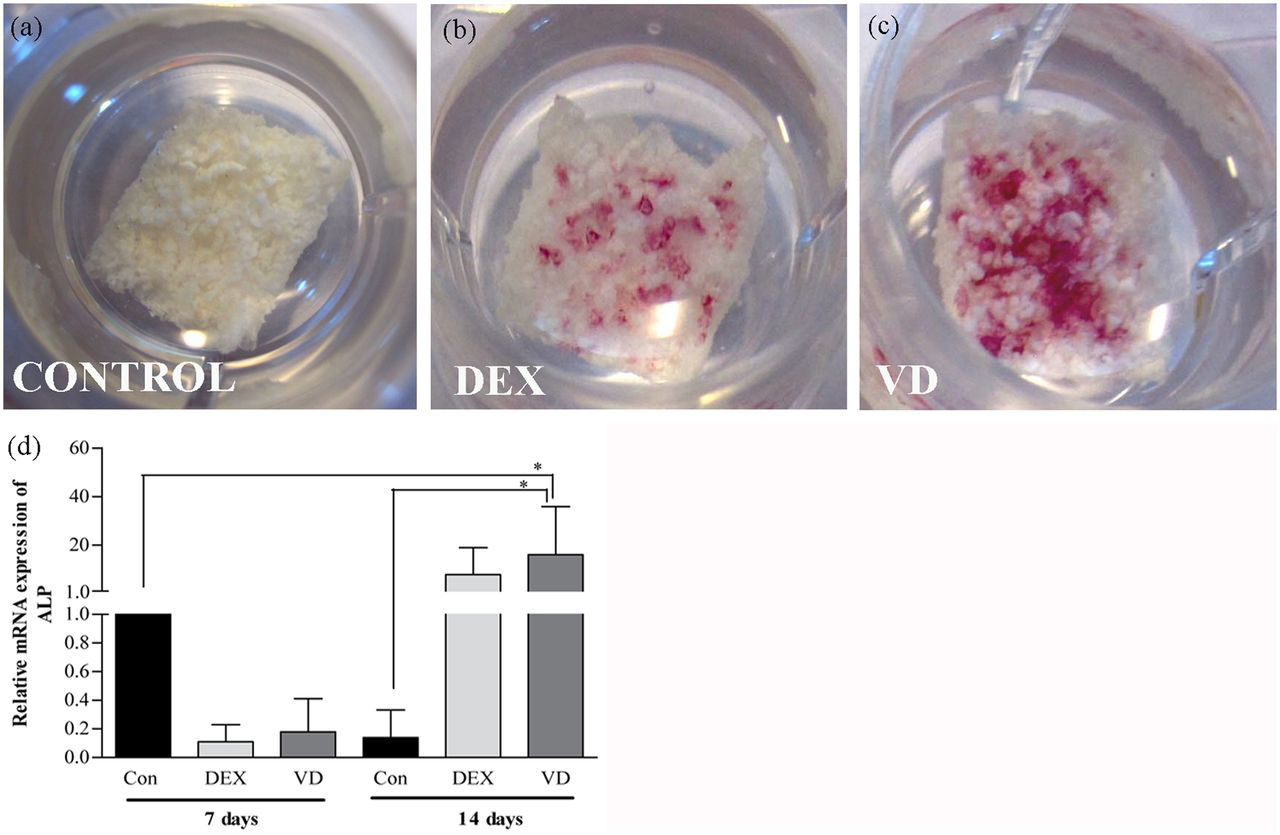
ALP staining and relative mRNA expression of ALP in DPSCs seeded on β-TCP/P(LLA/CL) 3D scaffolds cultured in (a) HS-M, (b) OM-DEX, and (c) OM-VD. Weaker ALP staining was observed in scaffolds cultured in (b) OM-DEX than (c) OM-VD at day 14. (d) The relative mRNA expression of ALP gene in DPSCs cultured in OM-VD was significantly higher than HS-M at days 7 and 14 time points. Results are reported as change in gene expression relative to untreated control (HS-M) set as 1 at day 7 time point.
mRNA expression and immunostaining for OCN
The deposition of OCN in the ECM was used as a marker of osteogenic differentiation of DPSCs within the biomaterial scaffold. OCN protein expression was assessed by immunostaining (Figure 7(a) to (c)), the cells cultured in OM-VD expressed OCN slightly more in comparison to OM-DEX. Moreover, cells cultured in HS-M did not express OCN. When OCN mRNA expression was assessed, the OCN levels were significantly higher (p < 0.001) in cells cultured in OM-VD at day 14 in comparison to OM-DEX (Figure 7(d)). Also, OCN mRNA expressions were significantly (p < 0.001, p < 0.001) upregulated, when the control samples at day 7 and day 14 were compared with OM-VD cultured cells at day 14.
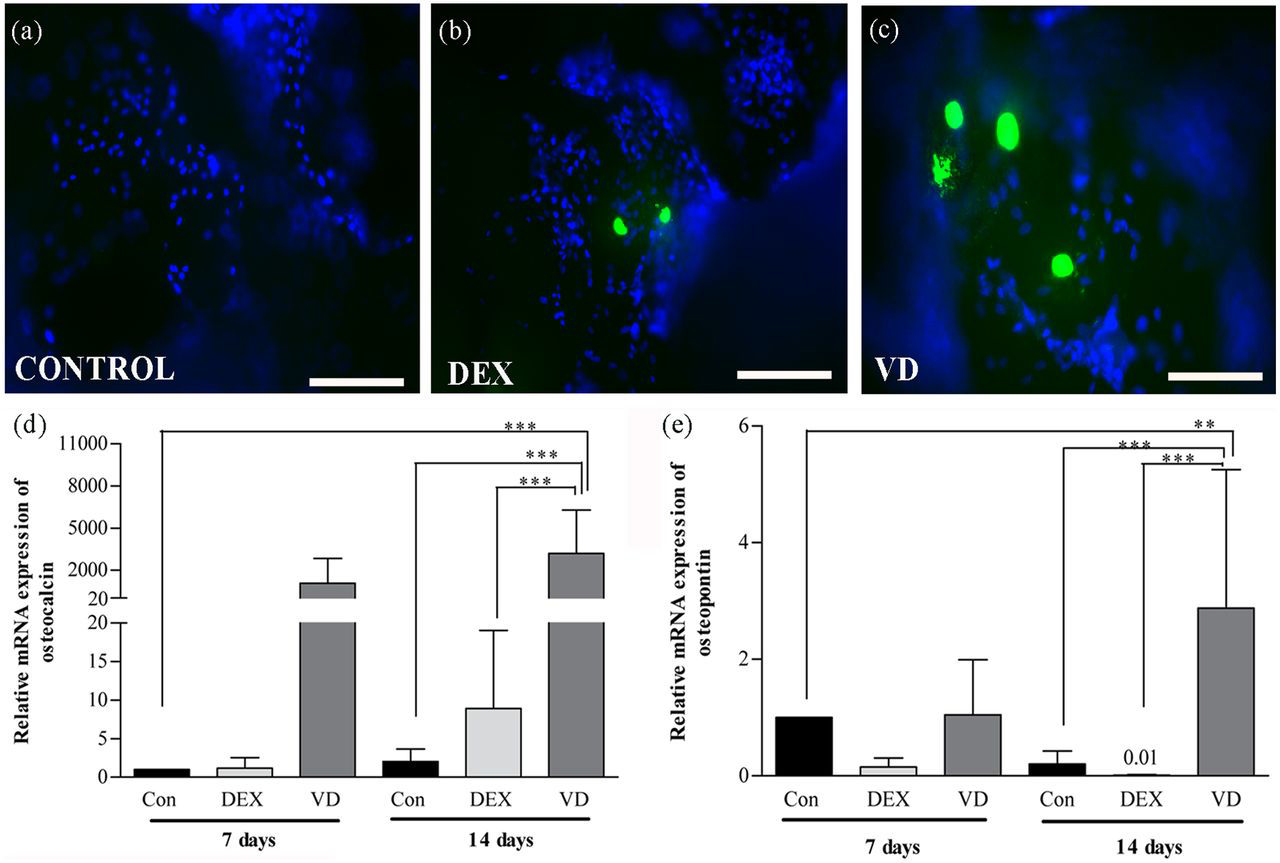
Representative immunofluorescence images of OCN expression in DPSCs seeded on β-TCP/P(LLA/CL) 3D scaffolds cultured in (a) HS-M, (b) OM-DEX, and (c) OM-VD (scale bar = 200 µm). The relative mRNA expressions of late osteogenic markers (d) OCN and (e) OPN in differentiated DPSCs cultured in OM-VD were significantly higher than untreated control (HS-M) and OM-DEX treated cells. Results are reported as change in gene expression relative to untreated control (HS-M) set as 1 at day 7 time point.
Consequently, we studied the expression of another late osteogenic marker, OPN, at mRNA level. Similar significant OPN expressions were observed as OCN in different treatment conditions, even though the relative mRNA expression levels of OPN were weaker than relative mRNA expression levels of OCN. OPN mRNA expressions were significantly (p < 0.01, p < 0.001) upregulated, when the control samples at day 7 and day 14 were compared with OM-VD cultured cells at day 14 (Figure 7(e)). In addition, at day 14, cells treated with OM-VD significantly (p < 0.001) upregulated OPN levels when compared with cells cultured in OM-DEX.
Discussion
Reconstruction of the alveolar bone by autografts or allografts to place dental implants requires an invasive technique to harvest grafts with a number of disadvantages including donor site morbidity, compromised vascularity, or limited tissue availability. 36,37 Present treatment modalities for bone regeneration by tissue engineering necessitate temporarily substituting ECM by the use of synthetic porous, osteoconductive biomaterials that would assist in cell attachment and osteogenesis. 25,29 The natural biomaterials such as chitosan and cellulose have certain disadvantages including low mechanical stiffness, lot-to-lot variability, and may be prone to contamination. 38,39 Further considering the clinical application of tissue-engineered bone grafts, the use of animal-derived components are not recommended, because they can elicit an immune response in patients upon implantation. 21 This is because human cells are able to take up animal proteins and present them on their membranes, thereby initiating xenogenic immune response leading to failure of the tissue-engineered graft. 20,40 Therefore, our research concentrated on xeno-free alternatives for animal-derived culture reagents and use of medical-grade synthetic composite material for bone tissue engineering as a step toward translation to clinical application. Here, we have described the viability, cell attachment, proliferation, and osteogenic differentiation ability of the DPSCs within a synthetic biomaterial β-TCP/P(LLA/CL) 3D scaffold maintained in xeno-free conditions.
Notably, to our knowledge, this is the first study where DPSCs were maintained in xeno-free culture conditions in vitro. In the literature, there are abundant reports discussing the isolation and culture condition for DPSCs. 11,15,41 Nonetheless, in these studies, DPSCs were cultured in FBS, which adds to the risk of transferring xenogenic antibodies and transmitting viral/prion diseases upon clinical application. 19,42,43 Moreover, HS has been explored as a possible alternative to FBS for culturing MSCs. 23,44 Furthermore, it was reported that HS could support proliferation and differentiation of human MSCs as efficiently as FBS medium in vitro and could maintain their bone forming capacity in vivo. 22 In this study, DPSCs cultured in HS adhered to the cell culture plastic and expressed membrane molecules CD90 and CD105 showing the mesenchymal origin of the cells and lacked expression of the hematopoietic markers CD31 and CD45, fulfilling the minimum criteria for defining MSCs. 45
Furthermore, the clinical outcome is influenced by many different factors. For successful initial approach toward bone tissue engineering, the interaction between potential osteogenic MSCs, osteoconductive biomaterial, and differentiation factors maintained in xeno-free conditions are the most relevant factors to be considered. Our choice of MSCs derived from dental pulp tissue was based on their ability to proliferate and differentiate osteogenically. Additionally, DPSCs are readily accessible and involve no invasive technique to obtain MSCs, which makes them a good candidate for use in bone tissue engineering. In a recent study, it was shown that canine DPSCs in combination with platelet-rich plasma formed mature bone with neovascularization and the response was similar to that of canine BM-MSCs at the dental implant site. 46 Apart from that, the ability of human DPSCs to regenerate into bone has been reported in several in vitro/in vivo studies 18,42,43 and also in a clinical study. 19
In our study, we have shown that DPSCs are attached, remained viable, proliferated, and differentiated osteogenically within β-TCP/P(LLA/CL) scaffolds. With the purpose of distribution of cells and osteoconductivity within a biomaterial, porous β-TCP with good bone bonding properties is preferred. 27 But due to poor mechanical strength, β-TCP is used for bone regeneration at nonloading sites or to fill bone defects. 28 From a tissue engineering point of view, a biomaterial should have sufficient strength initially to withstand the stresses of mastication until the newly regenerated bone takes over. 47 Moreover, the structural integrity is also crucial for the long-term success of implants in the bone. 4 In order to achieve desirable mechanical strength for bone tissue engineering, synthetic polymer–based biomaterials such as PLLA and PCL are combined with osteoconductive bioceramics. 30,48 With respect to biomaterial properties, β-TCP/P(LLA/CL) 3D biomaterial scaffold was preferred for our study; it influenced the adhesion of DPSCs onto the biomaterial surfaces and supported osteogenic differentiation of the cells within the porous solid structures.
To our knowledge, this is a first study showing the osteogenic potential of DPSCs within β-TCP/P(LLA/CL) 3D biomaterial scaffolds induced by OM-VD or OM-DEX. Having shown in our previous study that OM-VD supported osteogenic differentiation of the DPSCs better than the OM-DEX medium in vitro, we next sought to evaluate the response of osteogenic media on DPSCs within the β-TCP/P(LLA/CL) scaffolds. 14 Consequently, to assess the differentiation of DPSCs toward osteogenic lineage, ALP staining and expression were studied. ALP is a known marker for detection of early osteogenic differentiation of cells and is an ectoenzyme involved in the degradation of inorganic pyrophosphate to release phosphate for mineralization. 35 Importantly, in our study, we observed that DPSCs seeded in the scaffolds and cultured in OM-DEX and OM-VD expressed ALP activity within the biomaterial. Nevertheless, DPSCs induced with OM-DEX showed weak ALP activity, and the pattern was quite similar in all three patient cell samples. In addition, mRNA expression of ALP was observed to be significantly increased in the cells cultured in OM-VD. At the same time, the expression of OCN which is considered a late osteogenic marker 49 was evaluated, the levels were upregulated when the cells were treated with VD OM. Moreover, it is known that OCN is an important osteogenic marker that regulates the formation of mineral nodules and hence leads to osteogenesis. 50 Therefore, the expression levels of OCN are commonly correlated with the mineralization ability of the cells. Previously, weak levels of OCN have been reported in cells treated with OM-DEX. 51,52 Similar findings were observed in our study. Another important late-stage osteoblast differentiation maker OPN 53 was expressed when cells were cultured in VD OM. Considering the mRNA expressions of the genes, our results indicate that there was osteogenic differentiation, and ALP staining results showed that there was matrix mineralization in vitro. Based on our findings, β-TCP/P(LLA/CL) scaffolds strongly supported the osteogenic differentiation of DPSCs induced by VD OM. However, weaker osteogenesis was observed by OM-DEX in comparison to VD OM.
In summary, from our results, we can conclude that human DPSCs have the ability to survive, proliferate, and differentiate into osteogenic lineage within β-TCP/P(LLA/CL) scaffolds in vitro, which is important before evaluating the efficacy in vivo. Furthermore, the animal-derived cell culture supplements such as FBS was replaced with xeno-free supplements and medical-grade synthetic composite biomaterial was used. Hence, the outcome of this study can be directly applied to perform future clinical trials.
Footnotes
Acknowledgements
The authors thank Synthes for kindly providing the biomaterial for our study. The authors also thank Minna Salomäki, Anna Maija-Honkala, Miia Juntunen, and Sari Kalliokoski for their excellent technical assistance.
Funding
This study was supported by Finnish Funding Agency for Technology and Innovation (TEKES) and Competitive Research Funding of Pirkanmaa hospital district (9L057, 9M058).