
Abstract
An exogenous supply of growth factors and bioreplaceable scaffolds may help bone regeneration. The aim of this study was to examine the effects of TGF-β1 and VEGF-A transgenes on the osteogenic potential of bone marrow stromal cells. Rat bone marrow stromal cells were transfected with plasmids encoding mouse TGF-β1 and/or VEGF-A complementary DNAs and cultured for up to 28 days. Furthermore, collagen scaffolds carrying combinations of the plasmids-transfected cells were implanted subcutaneously in rats. The transgenes increased alkaline phosphatase activity, enhanced mineralized nodule formation, and elevated osteogenic gene expressions in vitro. In vivo, messenger RNA expression of osteogenic genes such as BMPs and Runx2 elevated higher by the transgenes. The data indicate that exogenous TGF-β1 and VEGF-A acted synergistically and could induce osteoblastic differentiation of bone marrow stromal cells in both cell culture and an animal model. The results may provide valuable information to optimize protocols for transgene-and-cell-based tissue engineering.
Introduction
Osteoblastic differentiation is essential for bone healing; the regenerative process involves the continual generation of osteoblasts and bone formation after the terminal differentiation of these cells. The supply of osteoblasts is sustained by an abundance of mesenchymal stem cells and intermediate osteoprogenitor cells. 1,2 However, the cell repositories, which can elicit cell proliferation and differentiation important for osteogenesis, are still to be defined. The reservoir of mesenchymal stem cells and osteoprogenitor cells may lie within the bone marrow, periosteum, and muscle connective tissue, 3 –7 for instance, cells derived from these tissues mineralize when cultured in vitro in the presence of ascorbic acid, β-glycerol phosphate, and dexamethasone 8 –10 and contribute to bone formation when transplanted into ectopic sites in vivo. Although the mechanism by which these cells proceed to osteoblastic differentiation remains unclear, they could play roles in normal bone homeostasis.
The use of endogenous stem or progenitor cells within predesigned matrix scaffolds along with cytokines or growth factors for local delivery has presented alternative strategies for more effective tissue engineering. 11,12 In particular, growth factors promoting spinal fusion have been increasingly investigated for their capacity to enhance bone regeneration. However, several barriers to effective and safe local delivery of such proteins in vivo have been identified 13 : their very short life, high production cost, and inability to maintain full bioactivity after their incorporation into controlled delivery systems. Moreover, high-dose local delivery can be associated with both local and systemic toxicities. Gene therapy is an effective approach by which therapeutic proteins can be delivered in a physiological and persistent manner. 14,15 One method of gene delivery is to use plasmids to carry cytokine and growth factor genes. Plasmid DNA possesses a stable and flexible chemistry and does not cause systemic toxicity because of the high efficiency of DNA catabolism in the bloodstream. 16
Transforming growth factor-beta 1 (TGF-β1), the largest source of which is bone, 17 has been implicated in osteoblast proliferation and differentiation 18,19 and is expressed at high levels during bone growth and development with an adequate blood supply. 17,20 –24 Vascular endothelial growth factor (VEGF), which is secreted by many cells including osteoblasts and osteoblast-like cells, plays an important role for adequate angiogenesis 25,26 and may be intimately related to bone development and fracture healing because both intramembranous and endochondral ossifications are associated with capillary development. 27,28 VEGF expression is regulated by several growth factors, hormones, and cytokines, such as TGF-β1. 29 These two proteins are associated with osteogenesis during bone growth, development, and healing; but they do not stimulate stem cells or bone progenitor cells to generate to be osteoblasts as directly as bone morphogenetic proteins (BMPs). However, these proteins have efficacy on not only cell migration and propagation but also on angiogenesis indispensable for bone formation. Furthermore, cells stimulated and proliferated by these growth factors could produce and secrete more BMPs to osteoblastic progenitor cells and preosteoblasts for osteoblastic maturation. Although many growth factors including TGF-β1and VEGF have been examined in vitro and in vivo in studies of cellular or molecular events, the combination of TGF-β1 and VEGF, especially in vivo, has rarely been investigated.
In this study, we aimed to examine the effects of TGF-β1 and VEGF-A transgenes on the osteogenic potential of bone marrow stromal cells in culture and in implanted bioreplaceable scaffolds in vivo. We introduced transgenes of mouse TGF-β1 (mTGF-β1), mouse VEGF-A (mVEGF-A), and an mTGF-β1/mVEGF-A combination in rat bone marrow stromal cells by lipofection using plasmid DNA and investigated their effects on cell proliferation, osteoblastic differentiation, and osteogenic gene expressions.
Materials and methods
The study protocol was approved by the institutional Recombinant DNA Experiment Committee and Animal Experiment Committee. All the experimental procedures were performed in accordance with the institutional guidelines for the care and manipulation of laboratory animals.
Plasmids encoding mTGF-β1 and mVEGF-A genes
pGT-mTGF-β1 plasmid (7888 bp) and pGT-mVEGF-A plasmid (7362 bp), encoding mTGF-β1 complementary DNA (cDNA) (570 bp; full length) and mVEGF-A cDNA (1170 bp; full length), respectively, were purchased from Invitrogen. Each plasmid included the hEF1-HTLV promoter for transcription of the genes in the transfected cells. The plasmids were adequately cloned and purified by using a Plasmid Purification Kit (QIAGEN).
Harvest and culture of bone marrow stromal cells
Bone marrow stromal cells were sourced from male Sprague Dawley rats (400 g). After the animals were euthanized in a CO2 chamber, their femurs were resected and bone marrow stromal cells were obtained from the distal femurs by flushing the marrow cavities. The cells were seeded in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin in 75-cm2 flasks. Then, only the primary culture medium received 1.25 µg/mL amphotericin B (Fungizone; Gibco-BRL). The cells were repeatedly trypsinized and passaged in DMEM containing 10% FBS and 1% penicillin–streptomycin while growing confluent. At the fourth passage, the cells were plated at 1 × 105/well in 24-well plates.
Plasmid-based gene transfer
The plated cells were divided into four groups according to the type of transgene: the mTGF-β1 and mVEGF-A transgenic groups received the pGT-mTGF-β1 and pGT-mVEGF-A plasmids, respectively; the mTGF-β1/mVEGF-A transgenic group received a combination of the pGT-mTGF-β1 and pGT-mVEGF-A plasmids; and the nontransgenic (control) group received a vehicle vector. Both the plasmids encoded the enhanced green fluorescent protein (EGFP) gene, which is transcribed by the human cytomegalovirus immediate early promoter and first intron A (hCMV-IA) promoter and translated to the protein in the cytoplasm. After 24 h of culture, when the cell number reached semiconfluency, the medium was replaced with fresh one with no antibiotics, followed by lipofection with 1 µg plasmid carrying the mTGF-β1 or mVEGF-A gene, or with 2 µg plasmid of the combination (1 µg each) using Lipofectamine (Invitrogen). The cells trypsinized at days 1, 2, 4, 7, 14, and 28, were suspended in 100 µL phosphate-buffered saline (PBS) containing 0.1% t-octylphenoxypolyethoxyethanol (Triton X-100; Sigma) in 1.5 mL eppendorf tubes under ultrasound, and centrifuged. The supernatant was directly processed for EGFP measurement at 488 nm (excitation) and 509 nm (emission).
Alkaline phosphatase-positive cell staining and mineralized nodule staining
Naphthol AS-MX phosphate (0.1 mg/mL) and fast blue BB salt (0.6 mg/mL) were dissolved in Tris-HCl buffer (0.1 M, pH 8.8) containing N,N-dimethylformamide (0.5%) and MgCl2 (2 mM) and filtrated to serve as the alkaline phosphatase (ALP)-positive cell-staining solution. The cells trypsinized at days 7, 14, 21, and 28 were washed twice with PBS and fixed in 3.7% formalin for 10 min. After the fixed cells were rinsed with PBS twice, PBS was replaced with 1 mL of the staining solution and the cells were incubated at 37°C for 20 min until the ALP-positive cells stained blue. The staining reaction was stopped by washing with PBS. Digital images were captured with a microscope.
Then, the same cells were used for examining mineralized nodule. The staining solution in this case was prepared by dissolving alizarin red S (1%) in 1/100 water-diluted ammonium hydroxide and filtration. The cells were washed twice with PBS and immersed in methanol for 10 min. After tapping in water, the cells were incubated for 2 min with 500 µL of the staining solution per well until mineralized nodules appeared red. The staining reaction was terminated by washing with water. Digital images were captured with a microscope.
Fluorometric assay
Hoechst 33258 dye (Polysciences) was used for the assay to estimate the DNA amount. Cultured bone marrow stromal cells from three wells per group were trypsinized, suspended with 100 µL PBS containing 0.1% Triton X-100 (Sigma) in 1.5 mL eppendorf tubes under ultrasound, and centrifuged at days 1, 2, 4, 7, 14, and 28. The cell pellets were dissolved ready for chemical reaction in 2-mL Hoechst dye buffer (10 mM Tris, 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4). Then, 100 µL of each sample was distributed in a well of a 96-well plate, and 100 µL of Hoechst dye (1 µg/mL in Hoechst dye buffer) was added to each well. The fluorescence of the dye/sample complex was immediately measured at an excitation wavelength of 360 nm and an emission wavelength of 460 nm by a Bio-Tek FL500 fluorescence microplate reader (BioTek Instruments, Inc., USA).
Measurement of ALP activity
ALP yellow liquid substrate (Sigma) containing p-nitro-phenylphosphate was used for the assay. The cells were collected in the same manner at days 1, 2, 4, 7, 14, and 28 and directed to chemical reaction. Thereafter, 10 µL of each sample was placed in a well of a 96-well plate and 200 µL of the yellow substrate was added to each well. After incubation at 37°C in a dark chamber for 30 minutes, the reaction was terminated by adding 50 µL of sodium hydroxide (3 N). The yellow reaction product was read at 405 nm.
Effect of dexamethasone on mineralized nodule formation
Dexamethasone (10−8 M), β-glycerol phosphate (10 mM), and ascorbic acid (50 µg/mL) were added to the culture medium to serve as a conditioned medium for osteogenesis 24 h after lipofection. The conditioned medium was replaced with fresh medium every 7 days. At days 7, 14, and 28, the cultured cells were fixed in methanol and rinsed with deionized water. The mineralized nodules were stained with 1% alizarin red S (Sigma).
Real-time polymerase chain reaction analysis
The expressions of 15 genes related to osteogenesis were analyzed by real-time polymerase chain reaction (PCR). The primer pairs were designed by using Primer3 software (Table 1). Bone marrow cells from three wells per group were pooled and homogenized in TRIzol buffer (Invitrogen) at days 1, 2, 4, 7, and 14, followed by the extraction of total RNA using the SuperScript First-Strand Synthesis System for reverse transcription polymerase chain reaction (RT-PCR) (Invitrogen). The reverse-transcribed cDNAs were amplified by a Taq polymerase system with their compatible primer sets using the SmartCycler (Cepheid). The amounts of the PCR products during the PCR cycles were visualized as fluorescence curves of SYBR Green dye (Molecular Probes), which binds to PCR products in every cycle, and cycle threshold (Ct) was determined as the PCR cycle number at which the fluorescence reached the threshold. The expression of each gene at a time point was calculated relative to its expression in the control group at the same time point, after normalization to GAPDH expression in each group (Table 1). The expressions of the mTGF-β1 and mVEGF-A genes detected by real-time PCR analysis reflected the dilution of the carried plasmids during cell division.
PCR primer sets and formula to calculate relative amount of gene expression of interest to GAPDH gene expression

Fw: sequences of forward primer; Rv: sequences of reverse primer; Ta: annealing temperature.
2-(Ct-CtGAPDH); Ct: threshold cycle.
Implantation of mTGF-β1 and mVEGF-A transgenic bone marrow cells
As in the in vitro experiment, rat bone marrow stromal cells were obtained, cultured, divided into four groups, and transfected with the pGT-mTGF-β1 plasmid, pGT-mVEGF-A plasmid, combination of the pGT-mTGF-β1 and pGT-mVEGF-A plasmids, or a vehicle vector. The prepared cells were cocultured with collagen sponges (BD 3D Collagen Composite Scaffold; BD Biosciences), while being gently rotated to allow even distribution of the cells inside the scaffolds, in 5 mL DMEM with 10% FBS and 1% penicillin–streptomycin overnight. Then, the matrices of the four transgenic groups along with those of a group using a collagen sponge without cells were subcutaneously implanted in the backs of Sprague Dawley rats (4–6 months old). After the specimens developed in the animals for 2 weeks, the neotissues from every four animals were excised and subjected to architectural assessment by microcomputed tomography (µCT), histological examination by toluidine blue staining, and gene expression analysis by RT-PCR, respectively.
Results
Detection of transfection
The intensity of EGFP fluorescence diminished time dependently and faded by day 7 (data not shown). Real-time PCR analysis represented the mTGF-β1 and mVEGF-A gene expressions from the pGT-mTGF-β1 and pGT-mVEGF-A plasmids, respectively, in all the transgenic groups, which attenuated quickly by day 7 (Figure 1).

Efficiency of the gene transfer.
Altered cell phenotype in culture
All the transgenic cells showed less ALP-positive staining intensities at days 7 and 14, which approached the control (no transgene) level by day 28 (Figure 2). Mineralized nodule formation was visible in all the groups at day 21 (data not shown) but was more prominent in all the transgenic cells, as shown in Figure 2. However, continuous loading of dexamethasone, β-glycerol phosphate, and ascorbic acid in the culture medium reduced the mineralization in all the transgenic cells by day 21 (Figure 3).

ALP-positive cell staining and mineralized nodule formation.

Effects of dexamethasone on the transgenic cells.
Fluorometric assay of DNA showed excessive proliferation of the transgenic cells by day 28, whereas proliferation of the control cells reached a plateau level at day 14 (Figure 4(a)). In terms of the ALP activity, all the transgenic groups had higher levels of activity at day 28, whereas the activity in the control group was higher at day 7, peaked at day 14, and decreased thereafter to a level lower than that in all the transgenic groups (Figure 4(b)).

Cell proliferation and ALP activity.
Osteogenic gene expressions
As seen in Figure 5, the expressions of most of the osteogenic genes were stimulated at day 2 or 4 after immediate inhibition at day 1. All the transgenes increased the gene expression levels of ALP, rat TGF-β1 (rTGF-β1), basic fibroblast growth factor (bFGF), bone morphogenetic protein 2 (BMP-2), receptor activator of nucleic factor kappa B (RANK), receptor activator of nucleic factor kappa B ligand (RANKL), and runt-related gene 2/core binding factor alpha 1 (Runx2/Cbfa1) by 10 times or more than the expressions in the control group by day 2 or 4. Although osteocalcin (OC) gene expression was stimulated by less than tenfold in the mVEGF-A transgenic group at day 4, its expression prominently increased in the mTGF-β1 and mTGF-β1/mVEGF-A transgenic groups at day 2. Osteopontin (OP) gene expression was generally inhibited by the transgenes except for strong elevation in the mTGF-β1 and mTGF-β1/mVEGF-A transgenic groups at day 4. Unexpectedly, BMP-4 gene expression was either upregulated or downregulated and affected in an opposite manner in the transgenic cells at days 2 and 4. The expression profile of insulin-like growth factor I (IGF-I) was similar to that of BMP-4 until day 4 but its expression level in all the transgenic groups was still slightly elevated at day 7. The gene expression level of rat VEGF-A (rVEGF-A) was not greatly affected except at day 1. Interestingly, the gene expression of collagen type II (COL-II) was extremely stimulated until day 7 in all the transgenic groups, whereas that of collagen type I (COL-I) had a completely opposite expression profile. Osteoprotegerin/osteoclastogenesis inhibitory factor (OPG/OCIF) gene expression almost countered RANK transcription over time. The expression levels of all the osteogenic genes converged to the control level by day 14.
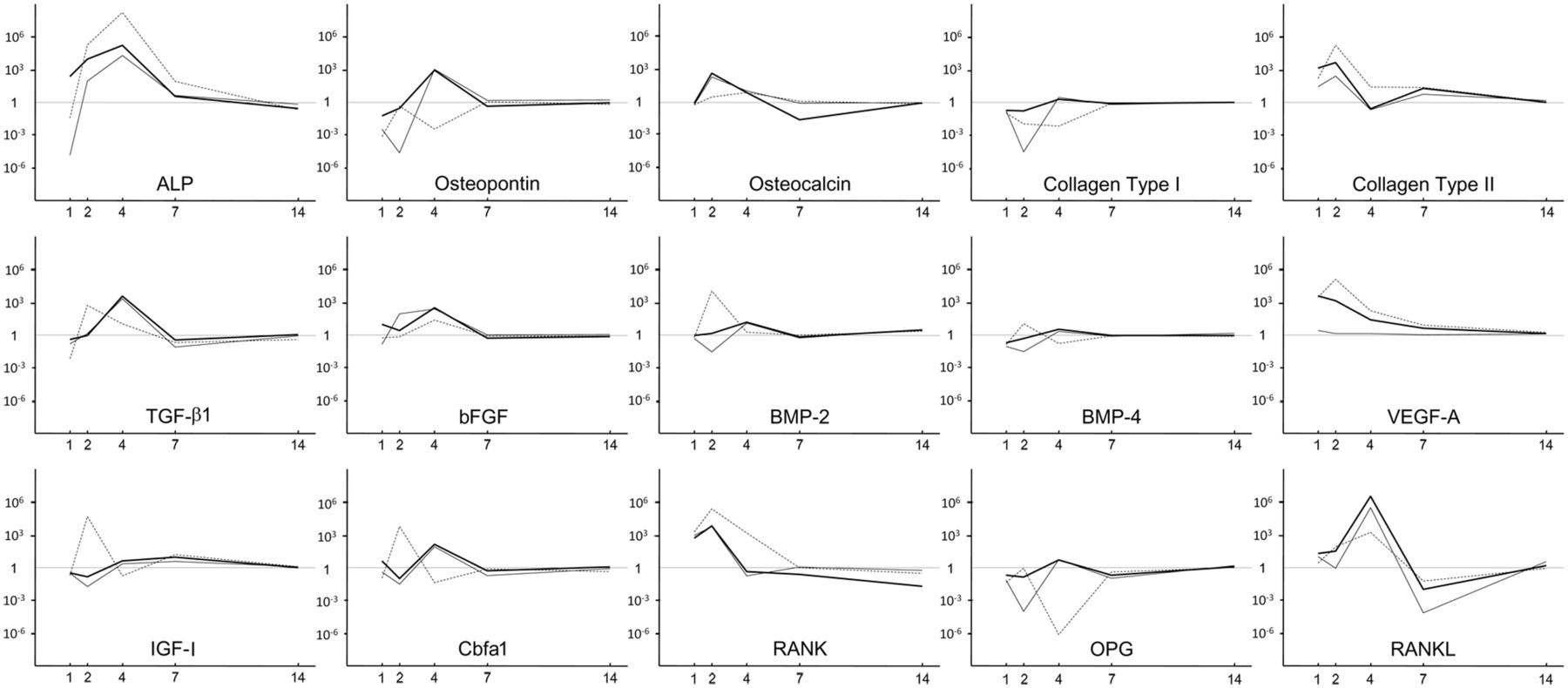
Profiling of the expressions of 15 osteogenic genes at days 1, 2, 4, 7, and 14 in vitro.
Composite implants
As represented in Figure 5, the expression levels of many osteogenic genes in the implanted collagen scaffolds increased in the mTGF-β1, mVEGF-A, and mTGF-β1/mVEGF-A transgenic groups (Figure 6). Especially, the mTGF-β1/mVEGF-A transgenic group showed more than tenfold gene expression levels of ALP, BMP-2, BMP-4, RANK, and Runx2 compared with the control group. However, the expressions of ALP, OC, and COL-II were downregulated in the mTGF-β1 and mVEGF-A transgenic groups, with COL-II showing the greatest response. rVEGF-A gene expression was downregulated in all the groups by more than sixfold. The µCT analysis did not bring quantitative information of bone volume over tissue volume (BV/TV) and other histomorphometric parameters among the groups at day 14 (data not shown) because of contraction and unevenness of the samples, although new mineral apposition was observed in all the groups (Figure 7); however, paraffin-embedded histological sections demonstrated greater cell invasion within the residual fibrous tissue in the three transgenic groups (Figure 8).

Real-time PCR analysis of the 15 osteogenic genes at day 14 in vivo.
An µCT image of a collagen sponge.

Histological view of the collagen scaffolds with the transgenic cells.
Discussion
This study demonstrated the effects of external application of TGF-β1 and VEGF-A as transgenes in a rat bone marrow stromal cell culture and in vivo. The exogenous messenger RNA (mRNA) expressions could be distinguished from the endogenous rTGF-β1 and rVEGF-A gene expressions by the use of specific primer sets. The method for gene transfer was lipofection with plasmid vectors encoding the mTGF-β1 and mVEGF-A genes, and it was interesting to clarify how transcription of the encoded genes would be sustained and influence molecular or cellular events. Chemical-based transfection represented by lipofection has been outstripped by viral methods in terms of transfection efficacy and sustained release of the translated proteins. 30,31 However, lipofection may be valuable as a nonviral method ensuring safety and ease of application, because chemical-based techniques such as lipofection and Ca-P precipitation generally provide mild expression of exogenous proteins carried as transgenes and their rapid degradation in the blood stream during cell segmentation with less probability of oncogenesis.
To observe transcription of the mTGF-β1 and mVEGF-A genes, their expressions were analyzed quantitatively by real-time PCR. The expressions gradually disappeared within 7 days after transfection, coinciding with the fading of EGFP fluorescence (data not shown). Therefore, although the protein levels of mTGF-β and mVEGF-A were not examined because of very low specificity of their antibodies, which may also be susceptible to rTGF-β1/rVEGF-A, we consider that mTGF-β1 and mVEGF-A protein secretion decreased in the same manner as that of EGFP. These results suggest that the plasmid-based gene transfer into rat bone marrow stromal cells by lipofection could maintain the transcription and translation of the encoded genes for 7 days, followed thereafter by a gradual decrease.
On the other hand, the 15 osteogenic genes examined in the cell culture after the gene transfer had different expression profiles, gradually responding to the transgenes with a peak at about day 7. The gene-carrying concept used in this study was lipofection of plasmid vectors, which is known as a conventional method but does not function for continuous or long-term transcription as well as viral transfections do. Therefore, efficacy of the transgenes could only show positive or negative peaks for driving osteogenesis in the cell culture at an early time; however, it is hypothesized that this initiation to the cells derived from bone marrow might have affected osteoblastogenesis in an autocrine and an paracrine manners. Although plasmid-based gene transfer has poor efficiency and has been modified with biocompatible matrices in vivo, 14,15,32 –34 endogenous gene transcription could be maintained effectively for a couple of weeks in response to the exogenous genes in vitro. This result indicates that gene transfer can be considered and designed not only for the carried genes but also for a cascade of altered endogenous genes.
TGF-β enhances the proliferation and early differentiation of osteoblasts, which are the processes characterized by a high rate of collagen synthesis, but impairs their differentiation based on OC synthesis and mineralization of bone matrix. 35 VEGF-A regulates human osteoblast differentiation in addition to its effects on osteoblast migration and proliferation. 36 Another study has suggested that VEGF does not stimulate fetal bovine osteoblast proliferation but induces migration, parathyroid hormone (PTH)–dependent cyclic adenosine monophosphate (cAMP) accumulation, and ALP increase in this cell culture. 37 Although the bone marrow stromal cells used in this study were derived from a different species (i.e. rats), they showed significantly enhanced proliferation influenced by either TGF-β1 or VEGF-A, carried as transgenes, as clarified by the increase in DNA synthesis in the fluorometric assay.
Both the transgenes temporally stimulated the expressions of 15 osteogenic genes including ALP. However, these transgenes repressed ALP activity significantly at days 7 and 14 and also reduced the intensity of ALP-positive cell staining at the same time points, followed by recovery of both ALP activity and ALP-positive cell staining close to the control levels at day 28 as the DNA synthesis increased. Therefore, the transgenic treatments may have regulated a posttranscriptional process of the ALP gene. However, mineralized nodule formation became more prominent in all the transgenic groups by day 28. Consequently, either the mTGF-β1 or mVEGF-A gene might have extended the cell proliferation stage and delayed ALP expression; the increase in mineralized nodule formation might be attributed to a change in transcription and/or a posttranscriptional process of the transgenes, which are expected to promote and enhance osteogenic differentiation.
A recent study has demonstrated that recombinant TGF-β1 does not influence gene expression or protein production of OC and ALP but dramatically abolishes BMP-2-mediated OC gene expression and ALP activity, inhibiting the ability of BMP-2 to induce mineralization in murine cell lines. 38 On the contrary, we found that although TGF-β1 delivery in rat bone marrow stromal cell culture by using a plasmid-repressed ALP gene expression at day 1 and ALP activity until day 14, mineralized nodule formation was finally enhanced with increased ALP gene expression, recovery of ALP activity at day 28, and elevated OC gene expression at days 2 and 4. The enhanced mineralized nodule formation could also be explained by the upregulated expressions of other genes at the early time points (days 2–7), such as OP, bFGF, rTGF-β1, BMP-2, BMP-4, Runx2, and IGF-I.
Interestingly, our study showed that VEGF-A gene transfer also stimulated mineralized nodule formation. As previously reported, VEGF expression is regulated by several growth factors, hormones, and cytokines including IGF-I and TGF-β1. 29,39 However, the effects of VEGF on the induction of osteogenic gene expressions are still not clear. We noted that the mVEGF-A gene induced a similar change in most of the examined osteogenic genes as the mTGF-β1 gene, including IGF-I and rTGF-β1 stimulation at an early time point (day 2). VEGF gene transfection dramatically stimulated rTGF-β1, BMP-2, BMP-4, and IGF-I mRNA expressions in addition to rVEGF-A gene expression, which was also enhanced by TGF-β1 gene transfer in rat bone marrow stromal cells. Therefore, VEGF might contribute to the endogenous upregulation of gene expressions of TGF-β superfamilies, especially at early time points (days 2 and 4).
Signals downstream of VEGF and TGF-β interact and cross talk in a number of significant ways, acting either in a synergistic or antagonistic manner. 40 Although all genes profiled in this study were not responded by the transgenes in a consistent manner, the combination treatment demonstrated acceleration of osteogenesis: for example, some genes such as ALP, OC, and COL-II were downregulated by each gene but upregulated by the combination and others were influenced synergistically or in different patterns.
In the ossifying cultures, dexamethasone (10−8 M), β-glycerol phosphate (10 mM), and ascorbic acid (50 µg/mL) were continuously added to promote osteoblastic differentiation of rat bone marrow stromal cells following the transgenic treatments. Similar to a previous report, 41 our in vitro study showed that the set of osteogenic supplements in the medium at the given concentrations induced more mineralization than did the normal medium in the nontransgenic group; on the other hand, interestingly, all the transgenes dramatically inhibited mineralized nodule formation in the conditioned medium. Dexamethasone (10−8 M) with β-glycerol phosphate (10 mM) and ascorbic acid (50 µg/mL) increases ALP activity and collagen synthesis and induces abundant mineralization, 42,43 accompanied with high expression of BMP-2. 41 However, the negative effects of higher doses of glucocorticoids on bone include decreased bone formation, increased osteoclastic resorption, impaired calcium metabolism, and decreased production of sex steroids, 44 –46 whose direct effects include inhibition of cell cycle progression, 47,48 induction of apoptosis, 49,50 and impairment of osteoblast function through transcriptional and/or posttranscriptional inhibition of collagen, 51 Runx2, 52 and IGF-I and TGF-β. 44 Furthermore, recombinant TGF-β1 added to confluent cultures of fetal rat calvarial cells with ascorbic acid and β-glycerol phosphate inhibits the formation of bone nodules. 53 Taken together with our finding of increased rTGF-β1 and rVEGF-A gene expressions by the transgenes, enhancing mineralized nodule formation, we suggest the following: (1) the osteogenic growth factors might have been stimulated by supplying the appropriate dose of dexamethasone in the culture medium, although we did not determine the ideal dose to upregulate these growth factors; (2) however, the mTGF-β1 and/or mVEGF-A loading into the medium containing dexamethasone, ascorbic acid, and β-glycerol phosphate was so high that not only transcription but also ALP activity and mineralized nodule formation were inhibited in a counter effect.
In vivo gene expressions in the bioreplaceable collagen scaffolds were also affected by both the mTGF-β1 and mVEGF-A genes. Although the transgenes individually stimulated the expressions of several osteogenic genes, to less than tenfold, the fold change induced by the mTGF-β1/mVEGF-A combination was greater in many genes, suggesting that the dual-transgenic rat bone marrow stromal cells had upregulated expressions of genes involved in osteogenesis. Interestingly, the COL-I and COL-II gene expression levels showed the opposite response, which might indicate different roles of the transgenes in osteogenesis. Considering that the transgenes strongly stimulated RANK and RANKL gene expressions in the rat bone marrow stromal cells at the early time points until day 7 and greatly inhibited OPG/OCIF gene expression, TGF-β1 or VEGF-A gene transfer is likely to be an important tool not only for bone formation but also for osteoclastogenesis triggering bone remodeling. Although no difference in bone formation was detected among the groups by µCT at day 14, the altered gene expressions can be expected to cause bone regeneration at a later time point. Unfortunately, the carrier of collagen sponge in this study could not be maintained stable underneath the skin and was decomposed after a couple of weeks.
In conclusion, the results of this study demonstrate that exogenous TGF-β1 and VEGF-A applied as transgenes have mitogenic effects on and enhance osteoblastic differentiation of bone marrow stromal cells and act synergistically. The data on cell behavior and gene expression profiles provide valuable information to optimize protocols for cell-based tissue engineering.
Footnotes
Acknowledgements
The authors acknowledge Chris Hendrick and Aladino DeRanieri for their technical assistance in the cell culture and animal experiments.
Funding
This study was partially funded by NIH Grant AR42862 and the Grainger Foundation.