
Abstract
Background:
Anti-KS autoantibodies are rare myositis-specific autoantibodies that have been described to target asparaginyl-transfer RNA synthetase.
Methods:
Here, we review the published literature on critical issues concerning the detection of anti-KS antibodies and the clinical features associated with their presence.
Results:
Seven articles are reviewed, in all of which immunoprecipitation was employed for the detection of anti-KS antibodies. A total of 47 patients were included; the ratio of females to males was 1.9:1. In total, 46 (98%) of these patients had interstitial lung disease (ILD), which was the sole manifestation in half (50%) of them. Pulmonary pathology revealed 7 (27%) with usual interstitial pneumonia, and 16 (62%) with non-specific pneumonia. Arthritis was present in about one-quarter (26%) of patients, and the incidence of Raynaud’s phenomenon and mechanic’s hands was 19% and 32%, respectively. However, manifestations of myositis were rare (9%). In addition, three (11%) patients had malignant tumors. Most patients responded to glucocorticoid therapy.
Conclusions:
Identifying anti-KS in patients with ILD may be useful for treatment, but reliable practical detection is needed. Furthermore, clinicians need to be aware of the possible presence of anti-KS antibodies in patients with ILD, either isolated or in combination with myositis.
Keywords
Introduction
Idiopathic inflammatory myopathies (IIMs) are characterized by different degrees of inflammation of skeletal muscle and other organs, such as lung and skin. Anti-synthetase syndrome (ASS) is a rare, chronic autoimmune disease of undetermined cause, considered to be a sub-group of IIMs. 1 The hallmark of ASS is the presence of autoantibodies against aminoacyl-transfer ribonucleic acid (tRNA) synthetase, also known as anti-synthetase antibodies, or anti-ARS. 2 Anti-Jo-1 was the first such anti-ARS discovered. Many other anti-ARS antibodies were discovered later and even more have been discovered recently. Patients with this syndrome have a clinical picture that is characterized by the occurrence of a classic clinical triad encompassing myositis, articular involvement, and interstitial lung disease (ILD). Raynaud’s phenomenon, fever, and mechanic’s hands are also frequently observed.3,4 The severity and type of pulmonary involvement determines the prognosis of the disease. 4
Anti-KS autoantibodies presumably target asparaginyl-tRNA synthetase, and are found in part of a group of ASS seen in a few patients with IIMs.5 –12 Because of their rarity, there is very limited information available on the clinical presentation patterns and evolution of disease associated with anti-KS associated ASS or anti-KS syndrome. The aim of the present study was to review the published literature on anti-KS syndrome and summarize clinical features and prognosis.
Methods
The following search was performed in PubMed and Embase (up to May 2020): “anti-asparaginyl-tRNA synthetase OR KS autoantibodies” OR “Anti-KS”. After removal of duplicates and exclusion of articles based on article type (reviews and conference abstracts were excluded) and content (as based on reading the abstract) a full-text review was performed for all articles available in English. Additional articles were hand searched. The same patients described in more than one article, if explicitly stated, were only included once for a review of the associated clinical features.
Results
General information
A total of seven articles were included, six of which were reports from Japan (see Table 1). A total of 47 patients were included from 1999 to 2020. The ratio of females to males was 1.9:1, comprising 16 males and 31 female patients, with age of onset ranging from 24 to 75 years. KS antibodies were detected by RNA and/or protein immunoprecipitation (IP) in all of these studies.
Basic information on the patients with anti-KS autoantibodies as described in the literature.

B, black; F, female; IP, immunoprecipitation; M, male; W, white.
Clinical characteristics of the included patients
Through analysis of the published literature as shown in Table 2, it was concluded that the lung is the most frequently involved organ, such that 46 (98%) of the anti-KS antibody positive patients had ILD, and the initial clinical manifestations of most patients were dry cough, shortness of breath after exercise and other ILD symptoms. By further analysis, 23/47 (49%) patients were identified as having ILD as the sole manifestation during the whole course of the disease. Only one patient had a rash and myositis, and ILD was not detected in that individual.
Clinical features of patients with anti-KS antibodies, as described in the literature.
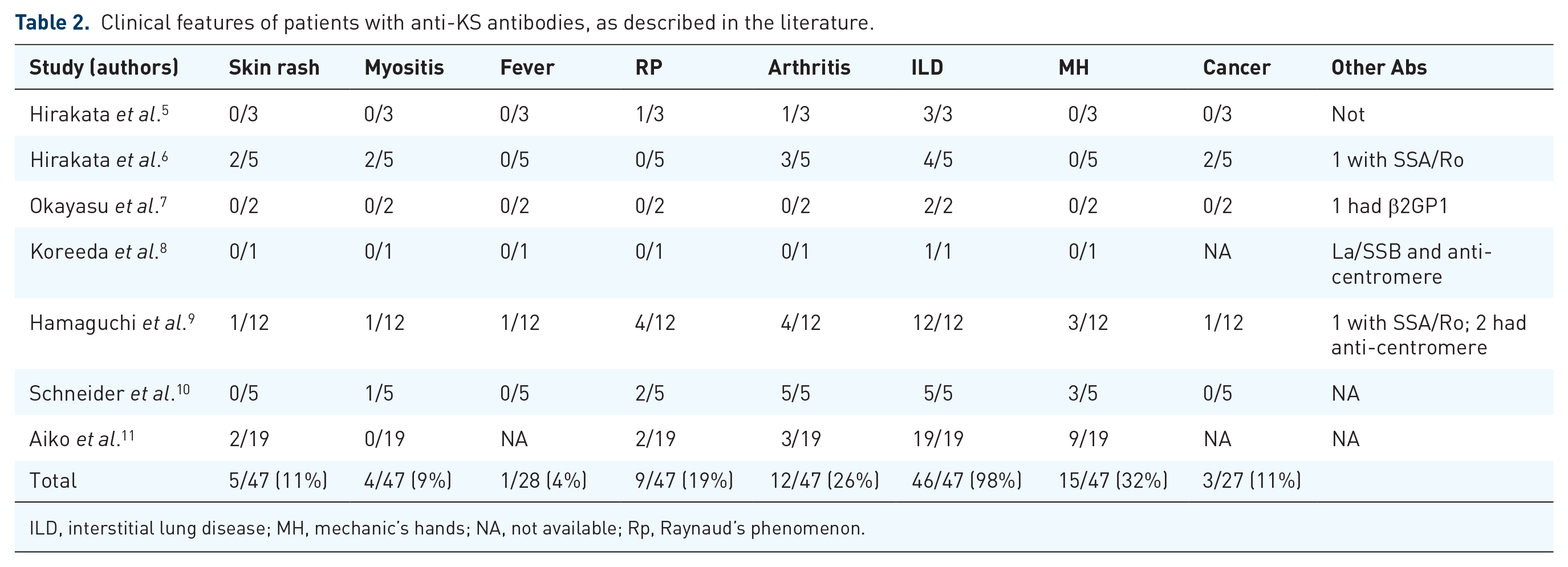
ILD, interstitial lung disease; MH, mechanic’s hands; NA, not available; Rp, Raynaud’s phenomenon.
Over the course of the disease, about a quarter (26%) of the patients showed signs of arthritis. The frequency of other clinical features was not high; Raynaud’s phenomenon was present in nearly one fifth (19%) of patients and mechanic’s hands were present in 15 (32%) patients, respectively. However, the incidence of skin rashes and muscle manifestations was only 11% (5/47) and 9% (4/47), respectively. Only one patient had symptoms of fever. According to the clinical characteristics, five patients can be diagnosed as having dermatomyositis (DM), including one patient with clinical amyopathic DM. We report that only 3% (1/39) of Japanese patients had myositis, while 43% (3/7) of European and American patients had muscular symptoms. During follow-up, three (11%) patients manifested or developed a malignant tumor. One was a 64-year-old Japanese patient who had prostate carcinoma prior to the anti-KS syndrome. In addition, one patient developed ovarian carcinoma 7 years after the onset of anti-KS syndrome, and another woman was found to have lung cancer after the onset of anti-KS syndrome. Pulmonary function was reported in 25 patients, mainly describing limited ventilation dysfunction; finally, pulmonary hypertension was reported in one patient.
Other autoantibodies were also detected in the serum of these patients with anti-KS antibody, including diverse antibodies such as anti-Ro/SSA, La/SSB, β2GP1 and anti-centromere antibodies. For three patients, anti-nuclear antibody (ANA) results were reported; these was only one ANA positive patient (1:80, homogeneous and speckled pattern). No patients had symptoms or indicators overlapping with other connective tissue diseases (CTDs).
Results of lung CR/CT and pulmonary biopsy histopathology
Imaging reflecting pulmonary changes was available from only 33 patients (Table 3). From the chest radiography (CR)/chest tomography (CT), it was determined that most of the patients have lung infiltration into both lobes, mainly at the base. Ground-glass opacity (GGO), reticulation, solid shadow, honeycomb shadow and other changes could be seen. Lung biopsies were performed by bronchoscopy, thoracoscopy, or thoracotomy in 26 of these patients. Pulmonary pathology was usual interstitial pneumonia (UIP) in 7/26 (27%) patients, non-specific pneumonia (NSIP) in 16/26 (62%), and organizing pneumonia (OP) in 1/26 (4%).
Pulmonary CT findings, pathological features, and treatment.
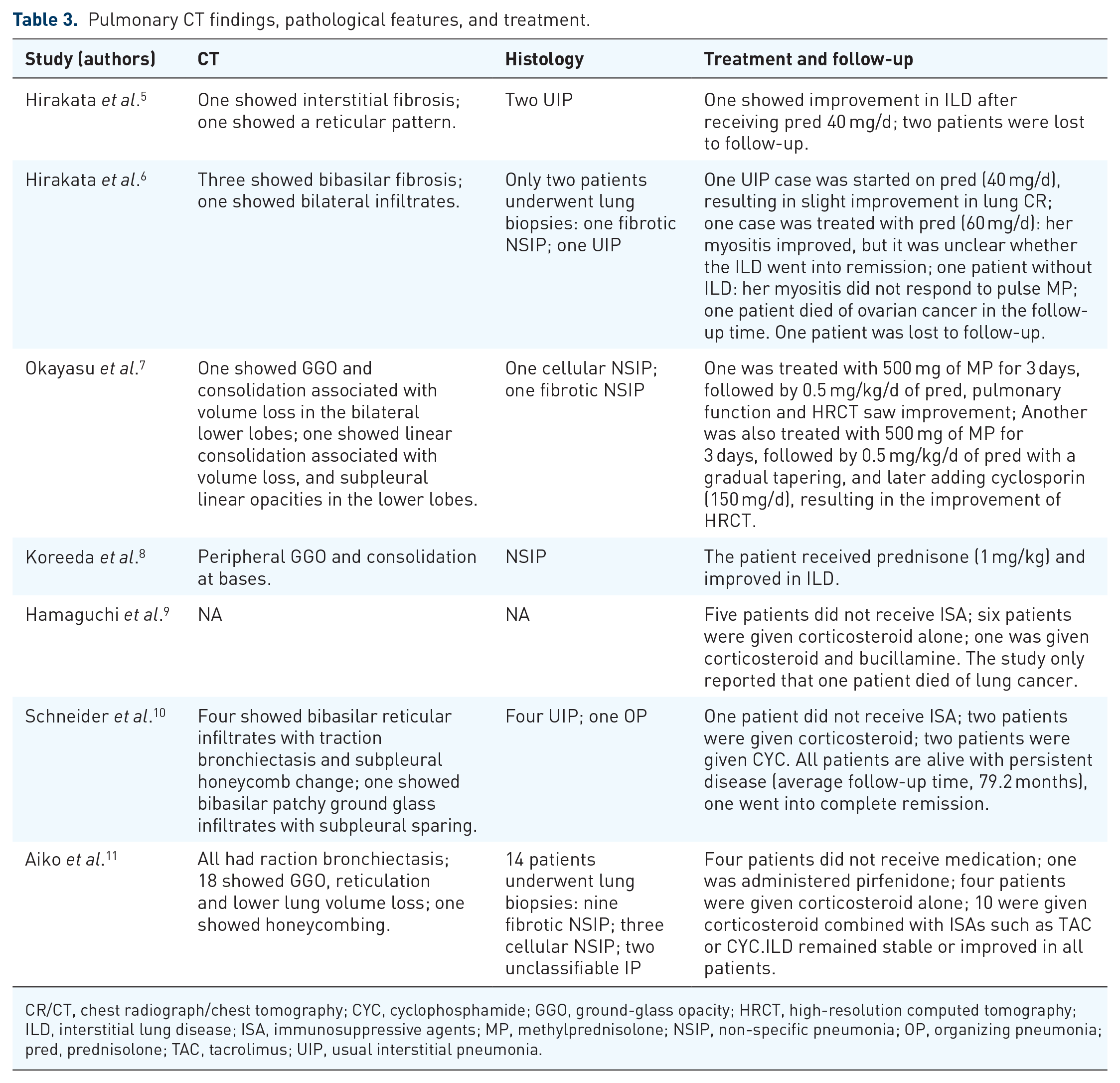
CR/CT, chest radiograph/chest tomography; CYC, cyclophosphamide; GGO, ground-glass opacity; HRCT, high-resolution computed tomography; ILD, interstitial lung disease; ISA, immunosuppressive agents; MP, methylprednisolone; NSIP, non-specific pneumonia; OP, organizing pneumonia; pred, prednisolone; TAC, tacrolimus; UIP, usual interstitial pneumonia.
Response to treatment
Two patients died of cancer during follow-up (one lung cancer and one ovarian cancer). In the reviewed literature, 11 patients who did not receive any immunotherapy continued to survive the disease during follow-up. Treatment options were reported for 32 patients, 18 patients of whom received glucocorticoid monotherapy including oral and/or intravenous administration, two received cyclophosphamide (CYC) treatment only, and the remaining 12 patients were given glucocorticoid combined with CYC, tacrolimus, cyclosporine, or bucillamine. The ILD improved or stabilized after treatment in 31 (97%) of these patients.
Discussion
In this review of the literature, anti-KS antibodies were reported as mainly detected by IP. Anti-KS syndrome was mainly seen in female patients with a chronic course, and the main manifestation was ILD. However, muscle involvement was seen in some patients, albeit rarely. Half of the anti-KS patients manifested ILD only, without any other symptoms. Anti-KS syndrome can be associated with tumors. The main pulmonary pathological changes were NSIP and UIP. Most patients responded well to treatment but the patients with tumors had a poor prognosis.
Many ARS have been identified, including anti-Jo-1, PL-7, PL-12, EJ, OJ, ZO, and WRS. 13 In 1999, Hirakata et al. reported that the same antibody was found in the serum of three patients named KS, KN, and NI. After further determination, it was identified as an antibody against asparaginyl tRNA, which was later uniformly designated “KS antibody.”5,12 Recently, an article retrospectively included 828 patients from 63 hospitals in order to establish the incidence of different ARS antibodies in ASS. This survey revealed the following degrees of prevalence: 593 anti-Jo-1 (72%), 95 anti-PL-7 (11.5%), 84 anti-PL-12 (10%), anti-38 EJ (4.5%), and 18 anti-OJ ARS (2%). 14 The frequency of anti-KS antibody in ASS patients varied from study to study, being less than 1% in the Hirakata et al. report, 5 while another study found that of 166 patients with anti-ARS antibodies, 13 (8%) did have anti-KS antibodies. 9 Because of the application of the IP method, the positivity rate seem lower than it actually is. IP remains the preferred detection method but is difficult to implement in daily practice for many hospitals. The need for practical and reliable immunoassays for detection of anti-KS is high because the detection of this antibody is relevant for diagnosis and treatment. However, it can be estimated that less than 5% of ASS patients possess the KS antibody.
In the present analysis of the literature, ILD was seen in 98% of those patients with anti-KS antibodies, and was in most cases the first definitive presenting clinical feature. ILD is one of the major features of ASS, and Raynaud’s phenomenon, mechanic’s hands, and arthritis, as seen in some anti-KS patients, are also felt to be part of the ASS. However, half of the anti-KS patients were suffering from ILD alone, and the incidence of ILD in the absence of other manifestations was higher than other types of ASS. 14 In previous studies, these ARS were associated with a high frequency of myositis (50%–84%) and arthritis (50%–90%), as well as an increase in Raynaud’s phenomenon (60%), fever with exacerbations (80%), mechanic’s hands (70%), and anti-Ro antibodies co-occurring in 50%–60% of cases.5,14 However, in this review article, we conclude that anti-KS syndrome could have characteristics that differ from those of other ASS because most of the patients with anti-KS antibodies showed late onset and a high incidence of ILD, together with a low incidence of myositis and low frequencies of Ro-52 antibody positivity. Therefore, it is likely that the clinical diagnosis varies among anti-ARS-based subgroups. The syndrome associated with anti-KS may be one end of the spectrum for ASS patients.
Hirakata et al. reported that anti-KS antibodies were found in 3% of Japanese patients with “idiopathic” ILD. 6 We are concerned that “idiopathic” ILD might be diagnosed without examination of anti-ARS antibodies because some patients with anti-KS antibodies show interstitial pneumonia without any symptoms suggestive of CTD. Although such patients have no apparent symptoms, interstitial pneumonia might be associated with autoimmune disease. This highlights the clinical importance of screening for such antibodies in patients with ILD even if there are no signs of myositis or CTD.
Based on the results of the chest high-resolution CT or lung biopsy, NSIP and UIP are the most common ILD patterns in patients with anti-KS antibody. The frequency of UIP in patients with anti-KS antibodies was relatively higher than expected from previous reports. Debray et al. reported that NSIP, OP, or mixed NSIP-OP patterns were observed in 45%, 21% and 24% of ASS patients, respectively. 15 Yamakawa et al. revealed that NSIP with OP overlap was present in 52.0%, NSIP in 46.7%, and OP in 1.3% of the ASS cases, and no patients had UIP in their cohort. 16 From the CT imaging and the main pathological pulmonary features, it is suggested that anti-KS syndrome may represent another discrete type different from other ASS.
In the report by Hirakata et al., it is noteworthy that the two patients with both anti-ARS and myositis were among the three patients from outside of Japan, while none of the five patients from Japan had myositis. 6 According to this analysis of the published literature, the incidence of myositis in Japanese patients is also much lower than in Western countries. This might suggest that patients with anti-KS antibodies in Western countries are more prone to muscle involvement, but this would need to be validated with a larger sample.
Malignancy has been reported to be unusual in patients with ASS. In the present review, 11% of patients with anti-KS antibodies were found to have malignancy during their disease course. During follow-up, two patients died, both of them from malignant tumors. This may suggest that malignancy is an important prognostic factor for this group. However, it is unclear whether the malignancies in these patients are related to anti-KS syndrome because they occurred separated in time from each other, in one patient even before the anti-KS syndrome, and one case as late as 7 years after the onset of the disease. More cases are needed to confirm any associations between the malignancy and the ASS disease.
In ASS patients, especially ASS-ILD, Ro is the most commonly detected antibody. One study revealed that Ro-52 antibodies were frequently detected among the anti-ARS (57% in Jo-1, 67% in PL-7), 17 but in the present review, less than 10% of the anti-KS patients were found to have Ro antibodies. Anti-KS syndrome might be a different entity from the other ARS.
Most of the patients with KS-ILD responded well after immunotherapy, but some of them were able to survive with ongoing disease even without immunotherapy. This seems to show that a chronic stable clinical course in patients with KS-ILD is a possible manifestation. In those patients with KS antibodies, in addition to the ILD, other symptoms consistent with the incidence of arthritis, Raynaud’s phenomenon, and mechanic’s hands, are more frequent than the incidences of myositis. This raises some doubt as to whether it is appropriate to describe anti-KS antibodies as myositis specific antibodies.
This review has several limitations. The study involves a relatively small number of patients at follow-up, and some of them were only followed for a relatively short period. Second, complete clinical data was not available for every patient.
In conclusion, this review integrated the published clinical, radiological, and pathological findings in, and treatment of, anti-KS syndrome. The data suggest that ILD is the main and most important clinical feature of patients with anti-KS antibodies. KS-ILD usually has a chronic clinical course, predominant GGO at the base of the lung, with volume loss, UIP and NSIP patterns, and a good response to corticosteroid treatment. KS-ILD may be a different disease entity from other types of ARS-ILD. It could be helpful to examine anti-KS antibodies in interstitial pneumonia even if signs of myositis or CTD are absent. Measurement of anti-ARS antibodies might be a clue to the possibility of autoimmune disease as a differential diagnosis of idiopathic NSIP or idiopathic pulmonary fibrosis. Further studies are needed to clarify the details of each KS-ILD.
Footnotes
Acknowledgements
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.