
Abstract
Fibrosis usually results from dysregulated wound repair and is characterized by excessive scar tissue. It is a complex process with unclear mechanisms. Accumulating evidence indicates that epigenetic alterations, including histone acetylation, play a pivotal role in this process. Histone acetylation is governed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDACs are enzymes that remove the acetyl groups from both histone and nonhistone proteins. Aberrant HDAC activities are observed in fibrotic diseases, including cardiac and pulmonary fibrosis. HDAC inhibitors (HDACIs) are molecules that block HDAC functions. HDACIs have been studied extensively in a variety of tumors. Currently, there are four HDACIs approved by the US Food and Drug Administration for cancer treatment yet none for fibrotic diseases. Emerging evidence from in vitro and in vivo preclinical studies has presented beneficial effects of HDACIs in preventing or reversing fibrogenesis. In this review, we summarize the latest findings of the roles of HDACs in the pathogenesis of cardiac and pulmonary fibrosis and highlight the potential applications of HDACIs in these two fibrotic diseases.
Keywords
Introduction
When responding to injury, damaged tissue begins the repair process to recover the tissue’s original architecture and function. This injury–repair process is complex and must be well orchestrated. Dysregulation of this process would result in fibrosis and ultimately organ failure. 1 Fibrosis can affect many organs, including the heart and lungs. 2 Despite the different pathways in the different processes of organ fibrosis, there are some core signal pathways, such as transforming growth factor β1 (TGF-β1), which is activated in virtually all fibrotic process. 3 TGF-β1 activates fibroblasts to become myofibroblasts. 4 Myofibroblasts are the key effector cells, characterized by the production of extracellular matrix (ECM) and the expression of alpha-smooth muscle actin (α-SMA). 4 The pathogenesis of fibrosis is unclear, for example, it is not completely understood why myofibroblasts are transient in the normal injury–repair process but are persistent in fibrotic diseases. 5 Increasing evidence reveals that epigenetic mechanisms play a pivotal role in this process and indicates that epigenetic methods may provide great therapeutic opportunities for this disorder.
Epigenetic process refers to the inheritable alterations in phenotype without changes in gene sequence, 6 which is critical in regulating gene expression. There are three major epigenetic modifications: DNA methylation, histone modifications, and microRNAs .7,8 Histone modification is a reversible process, which indicates the covalent posttranslational modification of histone proteins, including methylation, acetylation, phosphorylation, adenylation, ubiquitination, sumoylation, and ADP ribosylation. 9 The most common and best-characterized histone modification is acetylation. The acetylation of histone is governed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), 10 which work together to control histone acetylation (Figure 1A). HATs catalyze the transfer of acetyl groups from acetyl-CoA to the lysine ε-amino groups on the N-terminal tails of histones, which weakens the interaction of the histone tail and DNA, promoting transcriptional activation. In contrast, HDACs remove the acetyl groups from acetylated histones, increase chromatin condensation, and suppress gene transcription. 11

A. Histone acetyltransferases and histone deacetylases control histone acetylation. HATs transfer the acetyl groups to the lysine residuals on histone proteins; this weakens the interaction of histone and DNA to promote gene transcription. HDACs remove the acetyl groups on histone proteins, increase chromatin condensation, and suppress gene transcription. There are four major classes of HDACs, as listed.
Interactions between HATs and HDACs control the acetylation status, and the HATs and HDACs are usually linked or physically associated with other proteins that have catalytic activities 12 or are regulatory factors. 13 It is critical to consider their roles in a cellular context–dependent manner and their interactions with other regulators. 14 For example, inhibition of HAT p300 decreases the transcription of collagen and α-SMA in cardiac fibrosis by blocking the acetylated histone associated with the genes. 15 On the other hand, inhibition of HDAC induces general histone acetylation yet decreases collagen expression by depleted association of acetylated histone at the collagen promoter region. 16 The mechanisms of this change is not clear, but emerging data have shown that HDAC inhibition can induce histone deacetylation at a localized promoter to downregulate the target genes.16–19 In addition to histone, many transcriptional factors such as Sp1 and nuclear factor (NF)-kB are also targets of HDAC deacetylation.20,21 Inhibition of HDAC would also affect these proteins, which would affect the target gene transcription. A study in lymphoma cells observed increased transcriptional factor Sp1 acetylation by HDAC inhibition, which significantly decreased the binding of Sp1 to the Bcl-2 promoter region and contributed to Bcl-2 downregulation. 19 Thus, extensive evaluations of associated proteins that are involved in the transcriptional control of a target gene are needed to determine gene expression change when inhibiting HDAC.
HDACs are involved in many cellular processes. Dysfunction of the HDACs is associated with various diseases, such as cancer, diabetes, and cardiac hypertrophy.22–24 HDAC inhibitors (HDACIs) are molecules that bind with HDACs and interfere/block their functions. 25 HDACIs are involved in processes such as regulating gene expression and apoptosis, through acetylation of histone and nonhistone proteins.17,22 Currently, four HDACIs have been approved by the US Food and Drug Administration (FDA) for clinical use in hematologic tumors. 26 There are many clinical trials in progress of HDACIs for a variety of tumors 27 (Table 1). However, for many other diseases, such as fibrotic diseases, HDACIs are only in preclinical studies. In the past decade, a growing number of studies have demonstrated that HDACs are involved in the initiation and progression of fibrosis in multiple organs, including heart, 28 lung, 29 liver, 30 and kidney. 31 HDACIs have been shown to ameliorate various forms of fibrosis in animal models.16,32,33 In this review, we focus on the latest findings of the roles of HDACs in the pathogenesis of cardiac and pulmonary fibrosis and highlight the potential applications and limitations of HDACIs in these two fibrotic diseases.
HDACIs in clinical trials.

AML, acute myeloid leukemia; CTCL, cutaneous T-cell lymphoma; HIV, human immunodeficiency virus; MDS, myelodysplastic syndrome; PTCL, peripheral T-cell lymphoma.
Cardiac and pulmonary fibrosis
Cardiac fibrosis is the final stage of various cardiovascular diseases including hypertension, myocardial infarction (MI), and ischemic, dilated, and hypertrophic cardiomyopathy. 68 Excess deposition of ECM, such as collagen and fibronectin, impairs cardiac systolic and diastolic functions, disrupts electrical conduction, and predisposes to fatal arrhythmias, heart failure, even sudden death.69,70 Although our knowledge of cardiac fibrosis has advanced tremendously over the past decade, there are still no viable therapies to reverse this disorder. Similarly, a variety of lung injuries and diseases result in pulmonary fibrosis. 71 The most common and pernicious form of pulmonary fibrosis is idiopathic pulmonary fibrosis (IPF). 72 IPF is a chronic progressive disease with high mortality and unknown etiology. 73 The mechanisms governing fibrosis are not well understood, but studies show that epigenetic mechanisms are involved in the pathogenesis of cardiac and pulmonary fibrosis.74,75 HDACs participate in the process of cardiac and pulmonary fibrosis.76–78 HDACIs are reported to improve the resolution of these two kinds of fibrosis in preclinical studies .17,79 We reviewed the role of DNA methylation in organ fibrosis 80 ; here, we focus on HDAC family members in the process of cardiac and pulmonary fibrosis.
HDACs, HDACIs, and related clinical/preclinical studies
HDACs
Eighteen HDACs have been identified in mammals (Figure 1A) and are classified into four groups based on their homology to yeast proteins. 81 Class I includes HDAC 1, 2, 3, and 8, which exist in the nucleus. Class II has HDAC 4, 5, 6, 7, 9, and 10; these enzymes shuttle between the cytoplasm and nucleus, which further divide into two subgroups as Class IIa (HDAC 4, 5, 7, and 9) and Class IIb (HDAC 6, 10). HDAC11 is the sole member of Class IV, which shares the catalytic domain of both class I and II HDACs. These three classes of HDACs are all zinc-dependent enzymes, usually called classical HDACs. 82 However, Class III HDAC, including sirtuins 1–7, is nicotinamide adenine dinucleotide (NAD+) dependent. Sirtuins are most commonly correlated with aging. 83 We focus on the classical HDACs and their inhibitors in this review.
In addition to histone proteins, HDACs also deacetylate many nonhistone proteins to control diverse cellular processes. 84 As we mentioned earlier, some transcriptional factors are targets of acetylation, whose activities can be affected by HDAC or HDACI. 85 It is important to note that acetylation and deacetylation occur not only in the nucleus but also in the cytoplasm, and they influence the functions of target substrates. 84
HDACIs
In general, the zinc-dependent HDACIs have three pharmacophores: a Zn2+-binding group, a polar connection unit, and a surface binding or cap group. 86 Based on the structure of the Zn2+-binding group, the HDACIs are divided into four structurally distinct groups: (a) hydroxamic acids, (b) short-chain fatty acids (SCFAs), (c) benzamides, and (d) cyclic peptides. 87 The group of SCFAs is further categorized into four classes according to their distinguished structures: linear compounds, cyclic tetrapeptides, cyclic depsipeptides, and miscellaneous HDACIs.87,88
The first discovered HDACI was trichrostatin A (TSA), identified from nature sources. 89 Since then, numerous natural HDACI products have been obtained from microbes, dietary plants, and medicinal plants. 90 HDACIs have emerged as a new class of anticancer drugs; a series of HDACIs have been synthesized and the number of HDACIs is constantly being updated.
HDACIs target histone and nonhistone proteins. Many acetylated nonhistone proteins are deacetylated by HDACs 84 and can be modulated by HDACIs. 91 These nonhistone proteins include structure proteins (such as α-tubulin), chaperone proteins (such as Hsp90), DNA binding nuclear receptors (such as androgen receptor), transcriptional factors (such as p53), signaling mediators, and enzymes. 92 Therefore, HDACIs not only modulate the histone but also nonhistone protein acetylation statuses and affect their functions. For example, Quisinostat, a novel second-generation HDACI, increases p53 acetylation at K382/K373 sites, upregulates p21, and results in G1 phase arrest. 93 Panobinostat is a pan-HDACI that is approved for cancer treatment; its cytotoxicity is linked to α-tubulin acetylation. 94
Panobinostat is one of the four US FDA-approved HDACIs for clinical use, all of which are anticancer drugs. Panobinostat (Farydac®, Novartis) is a hydroxamic acid, a Class I and II HDACI, and was approved for treating multiple myeloma in 2015. 95 Belinostat (Beleodaq™, Spectrum) is a pan-HDACI that was approved for treating patients with relapsed or refractory peripheral T-cell lymphoma (PTCL) in 2014. 96 Romidepsin (Istodax®, Celgene) obtained FDA approval for treating cutaneous T-cell lymphoma in 2009 97 and then in 2011 was granted approval for treating PTCL. 98 Vorinostat (Zolinza®, Merck, also known as SAHA, suberoylanilide hydroxamic acid) was the first approved pan-HDACI for the treatment of T-cell lymphoma in 2006. 99
In addition to these HDACIs approved for clinical use, there are many clinical trials of HDACIs in other diseases34–67 (Table 1). SAHA was used in a phase II clinical trial to treat human immunodeficiency virus (HIV)–infected individuals 42 and in phase I/II clinical trials of sickle cell disease 36 with promising results. Givinostat (ITF2357), the hydroxamic acid HDACI, exhibited impressive therapeutic benefit and an excellent safety profile in a study in systemic-onset juvenile idiopathic arthritis. 100 Although there is no approved clinical trial on HDACI in fibrotic diseases, many HDACIs are under extensive investigation in preclinical fibrotic models, including cardiac and pulmonary fibrosis101–103 (Figure 1B). The preclinical studies of HDACIs in cardiac and pulmonary fibrosis are summarized in Table 2.
HDACIs used in cardiac and pulmonary fibrosis.
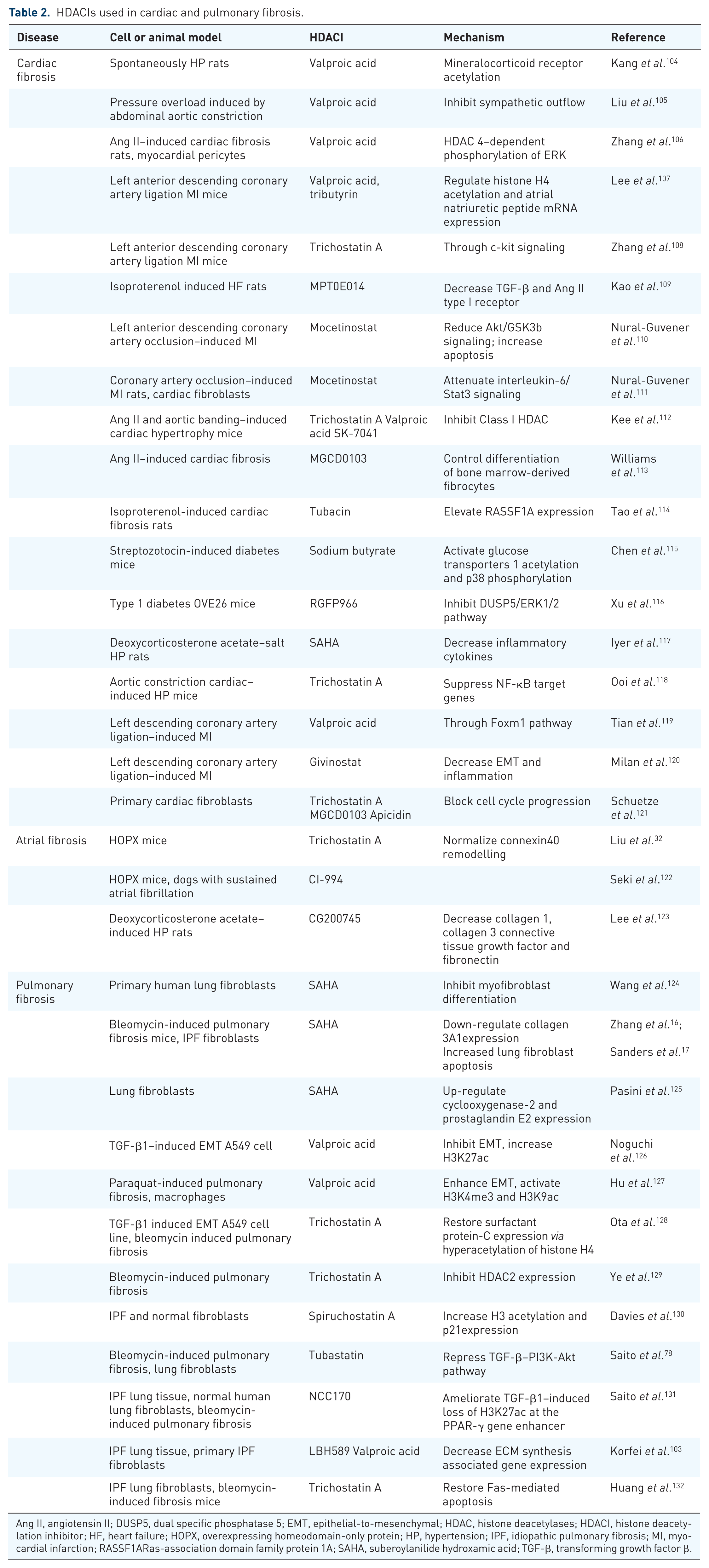
Ang II, angiotensin II; DUSP5, dual specific phosphatase 5; EMT, epithelial-to-mesenchymal; HDAC, histone deacetylases; HDACI, histone deacetylation inhibitor; HF, heart failure; HOPX, overexpressing homeodomain-only protein; HP, hypertension; IPF, idiopathic pulmonary fibrosis; MI, myocardial infarction; RASSF1ARas-association domain family protein 1A; SAHA, suberoylanilide hydroxamic acid; TGF-β, transforming growth factor β.
HDACs and HDACIs in cardiac fibrosis
Accumulating evidence has demonstrated that HDACs are dysregulated in cardiac fibrosis.133,134 Myocardial hypertrophy is both a pathological cause and a result of cardiac fibrosis. Initially, Class IIa HDACs were thought to act as endogenous inhibitors of cardiac hypertrophy. 135 The first connection between HDACs and cardiac remodeling was the finding that Class IIa HDACs interact with myocyte enhancer factor 2 (MEF2), a key regulator of myocardial hypertrophy. 136 Subsequent studies found that MEF2 interacts with HDAC4, HDAC5, and HDAC9 of Class IIa; overexpression of these HDACs decreases MEF2 expression and attenuates myocardial hypertrophy. 135 However, recent studies elucidated that Class IIa HDACs also exert profibrotic functions.28,133,137,138 For example, cardiomyocyte-specific HDAC4 overexpression promotes cardiac hypertrophy and exacerbates interstitial fibrosis in the model of MI in 6-month-old mice. 133 In another model of cardiac hypertrophy and fibrosis with natriuretic peptide receptor-A knockout mice, the HDAC7 expression is upregulated in parallel with an increase in TGF-β1 and collagen I. 28
In addition to Class IIa involvement in cardiac remodeling, genetic studies have suggested that Class I and IIb HDACs are also involved.76,134 Downregulation of HDAC1 and HDAC2 by gallic acid attenuated cardiac fibrosis in rat primary cardiac fibroblasts and in hypertensive mice. 134 HDAC3 was upregulated in an experimental model of heart failure; suppression of HDAC3 improved cardiac function and limited ventricular fibrosis. 139 HDAC6 activity was increased in deoxycorticosterone acetate–salt hypertensive rats and spontaneous hypertensive rats,76,140 and HDAC6 contributed to cardiac dysfunction and skeletal muscle wasting. 141
With the involvement of HDACs in the tissue remodeling process, many studies explored whether HDACIs could be used to correct the pathological remodeling in cardiac fibrosis. One of the major challenges in fibrotic disease is the increased myofibroblasts, the major effector cells of fibrosis. 4 Many preclinical studies have demonstrated that pan or selective HDACIs could reverse myofibroblasts activation, blunt myocardial hypertrophy, and preserve cardiac function in different myocardial hypertrophic and heart failure animal models.104,105,107,110 SAHA was reported to improve the sarcoendoplasmic reticulum Ca2+-ATPase activity in cardiac myocytes. 142 Some other pan-HDACIs also demonstrated their efficacy in cardiac fibrosis. Valproic acid (VPA)-attenuated cardiac hypertrophy and fibrosis in spontaneously hypertensive rats by affecting the mineralocorticoid receptor acetylation 104 ; it also prevented right ventricular hypertrophy in the rat. 143 In a study of a rat model of pressure overload, VPA inhibited sympathetic outflow and cardiac remodeling. 105 VPA was reported to ameliorate Ang II-induced pericyte-myofibroblast transdifferentiation and cardiac fibrosis through HDAC4-dependent phosphorylation of ERK. 106 VPA and tributyrin attenuated ventricular remodeling after infarction, likely through histone H4 acetylation and atrial natriuretic peptide mRNA expression in the cardiomyocytes. 107 A recent study demonstrated that VPA attenuated atrial remodeling and delayed the onset of atrial fibrillation in transgenic mice. 144 Other pan-HDACIs, such as MPT0E014, showed antifibrotic activities by decreasing TGF-β and Ang II type I receptor expressions in isoproterenol-induced dilated cardiomyopathy, 109 whereas TSA promoted myocardial repair and prevented cardiac remodeling via c-kit signaling. 108 TSA was reported to reverse atrial fibrosis and reduce the incidence of arrhythmia, without affecting the level of Ang II. 32 In a later study by the same group, the researchers used the same mice model, and a dog model of sustained atrial fibrosis demonstrated that Class I HDAC inhibitor CI-994 reduced atrial fibrillation and fibrosis. 122 These results provide evidence that HDACIs may be a new therapeutic option for atrial fibrillation.
In addition to pan-HDACIs, selective HDACIs have also demonstrated antifibrotic properties. The selective Class I HDACI, Mocetinostat, inhibited the upregulated HDAC1 and 2 in an animal model of congestive heart failure by reversing myofibroblast phenotype and increasing apoptosis. 110 Mocetinostat was also reported to attenuate interleukin (IL)-6/Stat3 signaling and decrease interstitial fibrosis and scar size in ventricular tissue in a rat model of heart failure. 111 In a study of cardiac hypertrophy, the Class I HDAC inhibitor SK-7041, similar to pan-HDACIs TSA and VPA, partially reversed pre-established cardiac hypertrophy. 112 MGCD0103 is another selective class I HDACI that inhibited Ang II-induced cardiac fibrosis by controlling the differentiation of bone marrow-derived fibrocytes. 113 Inhibition of Class I HDACs with Apicidin derivative was able to prevent cardiac hypertrophy and failure in preclinical studies. 145 HDAC6 of the class II HDACs was upregulated in cardiac fibroblast activation and fibrosis.76,140 Inhibition of HDAC6 by siRNA or its inhibitor, Tubacin, attenuated TGF-β1-induced myofibroblast markers, elevated Ras-association domain family protein 1A, and reduced cardiac fibrosis. 114
HDAC inhibition also has protective effects on the diabetic heart. Diabetes can induce severe cardiovascular complications including diabetic cardiomyopathy (DCM), a myocardial disorder without coronary artery disease by unclear mechanisms.115,116 Pan-HDACI sodium butyrate reduced interstitial collagen deposition and attenuated cardiac hypertrophy by mitigating apoptosis, increasing antioxidant SOD1, stimulating angiogenesis, and activating glucose transporters 1 acetylation and p38 phosphorylation in a streptozotocin-induced diabetic model. 115 Selective inhibitor RGFP966 of HDAC3 prevented fibrosis and the development of DCM by blocking the elevated phosphorylated ERK1/2, and upregulating dual specific phosphatase 5 (DUSP5) through increased acetylated histone H3 at DUSP5 promoter region in diabetic hearts. 116
In addition to the above-mentioned antifibrotic aspects of HDACIs, these inhibitors are reported to reduce inflammatory processes associated with fibrotic diseases. During fibrogenesis, including myocardial fibrogenesis, proinflammatory cytokines play an important role in fibroblast activation. 146 HDACs are regulators of inflammation and immunity. 147 Targeting proinflammatory cytokines exerts antifibrotic effects and improves cardiac function.117,119,120 SAHA was reported to decrease inflammatory cytokines, including IL-1α and vascular endothelial growth factor in deoxycorticosterone acetate–salt hypertensive rats. 117 VPA and Givinostat exhibited similar effects as SAHA in spontaneously hypertensive rats and an acute MI mouse model, inhibited proinflammatory cytokines production.119,120 These results demonstrate that HDACs are involved in inflammatory-mediated cardiac fibrosis.
Many studies with HDACIs demonstrated that these compounds exert overlapping and divergent effects. HDACIs are classified into different groups based on their chemical structure. 148 For example, TSA is a hydroxamic acid, MGCD0103 belongs to the aminobenzamides, while Apicidin is a cyclic peptide, 148 but they all present a common mechanism to inhibit the proliferation of cardiac fibroblasts and increase the expression of anti-proliferative genes to block cardiac fibrosis. 121 Yet these different HDACIs display different efficacy and effects, which should be carefully assessed for specifically targeted processes. A study in cardiac fibroblasts demonstrated that MGCD0103 but not TSA or Apicidin paradoxically increased fibrotic-related PAI-1 expression. 121 MGCD0103 and Apicidin are highly selective inhibitors of Class I HDACs, while TSA is mainly a Class I and IIb HDAC inhibitor. The divergent effects of these HDACIs are likely due to the different complexes that are specifically engaged with HDAC1 or HDAC2 by these inhibitors. 121
Many of these HDACIs are synthetic, but there are natural HDACIs, including caffeic acid and polyphenols, which have been shown to attenuate cardiac hypertrophy and myocardial fibrosis.149,150 It is worth mentioning that some natural compounds contain Resveratrol, the Class III HDAC sirtuins activator, could ameliorate cardiac fibrosis by activating sirtuin 3 and affecting the TGF-β/SMAD3 signaling pathway. 151
HDACIs have been reported to be beneficial for cardiac fibrosis in preclinical studies. Given the multiplicity of the distinct HDACs, it is critical to identify the specific functions of each HDAC to develop more selective and targeted inhibitors. Extensive studies are needed to understand the HDAC-mediated profibrotic processes in cardiac fibrosis, as HDACs may not only associate with the acetylation process in the cells. For example, HDAC2 can be phosphorylated, and the phosphorylation of S394 on HDAC2 would induce the binding of Hsp70 and result in cardiac hypertrophy in mice. 152 Epigenetic-based therapies are still limited in the cardiovascular field; more studies are needed to explore the usage of HDACIs in cardiac fibrosis, including its safety and long-term effects.
HDACs and HDACIs in pulmonary fibrosis
Pulmonary fibrosis is another devastating disease, with IPF as the most common form. Although there are newly approved drugs mainly to relieve symptoms, 73 there is no effective treatment. In the past decade, the involvement of HDACs in the pathogenesis of pulmonary fibrosis has been documented. The activities of all Class I and II HDACs were reported to be significantly upregulated in IPF lung tissues. 103 Immunohistochemistry demonstrated that nearly all of these HDACs had a strong induction in myofibroblasts at fibroblast foci and in abnormal bronchiolar basal cells at sites of aberrant re-epithelialization in IPF but not in the control lungs. 103 Among these HDACs, HDAC4 demonstrates its importance in lung fibrosis. HDAC4 can modulate the production of ECM in lung myofibroblasts; knocking down HDAC4 attenuates α-SMA expression stimulated by TGF-β1 in normal human lung fibroblasts. 153 HDAC4 is also involved in the early stress response, while HDAC2 is increased in the middle and late stages of bleomycin-induced lung fibrosis in mice. 102 A study with IPF fibroblasts showed that aberrant protein phosphatase 2A/HDAC4 axis suppresses miR-29, causing a pathological increase in type I collagen expression. 154 In addition to HDAC4, HDAC6 and HDAC8 expressions are reported to be altered in IPF.78,155
To target the dysregulated HDACs in pulmonary fibrosis, many studies used HDACIs to examine their effects in blocking the fibrotic process. Although studies involving HDACIs in pulmonary fibrosis are relatively limited compared with those in cardiac fibrosis, growing evidence highlights the antifibrotic effects of HDACIs in IPF. The most studied HDACI in IPF is SAHA, a nonselective HDAC Class I and II inhibitor. It has been reported that SAHA can inhibit the differentiation of TGF-β1-induced myofibroblasts 124 . It induces myofibroblasts apoptosis 17 and downregulates type III collagen expression on the transcriptional and translational levels. 16 SAHA also upregulates antifibrotic cyclooxygenase-2 (COX-2) protein expression and prostaglandin E2 (PGE2) production by posttranscriptional mechanisms. 125 Both SAHA and LBH589 (Panobinostat) have been reported to restore COX-2 expression in IPF.125,156
In addition to SAHA, other HDACIs have also demonstrated beneficial effects in lung fibrosis studies. VPA partially inhibited TGF-β1-induced epithelial-to-mesenchymal transition (EMT) process, in which VPA blocked the decreased histone acetylation (especially H3K27 acetylation) by TGF-β1 in human alveolar epithelial cells. 126 However, in a study in paraquat-induced lung fibrosis with macrophages, VPA aggravated the acute inflammation and worsened fibrosis partially through enhanced histone acetylation at the IL-6 promoter regions. 127 On the other hand, TSA attenuated bleomycin-induced lung fibrosis in mice; it restored decreased SFTPC, a gene encoding surfactant protein-C, via increased histone H4 acetylation at its promoter region in alveolar epithelial type II cells. 128 TSA upregulated the antifibrotic gene Thy-1 in rat lung fibroblasts 29 ; and through inhibition of HDAC2 expression, it reduced lung fibrosis in rats. 129
Studies on selective HDACIs also presented beneficial effects in pulmonary fibrosis. Spiruchostatin A (SpA), a selective HDACI, inhibited TGF-β1-induced α-SMA, collagen I, and collagen III expression. 130 Tubastatin, an HDAC-6 inhibitor, repressed TGF-β1-induced collagen expression by diminishing Akt phosphorylation. 78 However, this study demonstrated that in bleomycin-induced lung fibrosis in mice, Tubastatin-treated mice could be protected from lung fibrosis but not HDAC6 knockout mice. This indicates that Tubastatin may ameliorate lung fibrosis via an HDAC6-independent mechanism. 78 Inhibition of HDAC8 with its selective inhibitor NCC170 repressed TGF-β1-induced fibroblast–myofibroblast differentiation partially by increasing H3K27 acetylation at PPAR-γ enhancer regions to upregulate PPAR-γ mRNA, an antifibrotic molecule. 131 This inhibitor also ameliorated bleomycin-induced lung fibrosis in mice. 131
In addition to blocking the well-documented increased α-SMA and collagen expression in myofibroblasts, HDACIs have been reported to induce apoptosis by altering the apoptosis-related gene expression and subsequently alleviating fibrosis,17,103,132 as increased apoptosis resistance is a major characteristic of IPF myofibroblasts. 157 SAHA induces apoptosis in IPF primary fibroblasts, at least in part, by regulating apoptosis-related genes through epigenetic mechanisms. 17 Other HDACIs, LBH589 or VPA, considerably reduced the genes associated with the profibrotic and apoptosis-resistant phenotype in IPF fibroblasts. 103 Fibrotic fibroblasts with decreased Fas expression and increased resistance to Fas-mediated apoptosis are associated with increased HDAC 2 and 4; and TSA or SAHA can increase Fas expression in these cells and restore susceptibility to Fas-induced apoptosis. 132 Pirfenidone is the first anti-fibrotic agent for IPF to slow its progression, a recent study compared the antifibrotic efficacy of Panobinostat and Pirfenidone in IPF fibroblasts, and concluded that both have antifibrotic effects, but Panobinostat also induced cell cycle arrest and apoptosis and thus was more efficient in inactivating IPF fibroblasts. 158
HDACIs are powerful epigenetic regulators, and epigenetic mechanisms are involved in pulmonary fibrosis. 75 Many epigenetic factors that are affected by environmental changes, 159 including metabolic alterations 160 and aging, 161 contribute to pulmonary fibrosis. Histone modifications, especially histone acetylation, are only part of this complexed process. Inhibition of HDAC at in vitro and in vivo models of pulmonary fibrosis has shown promising results, but progress in determining the biological mechanisms and therapeutic options remains obscure. More in-depth studies in this area will empower us to develop better-targeted therapeutic methods to treat this fatal disease.
Limitations and side effects of HDACIs
HDACIs represent a new class of epigenetic drugs that have demonstrated promising results in preclinical studies. Histone and nonhistone proteins are the targets of HDACs, many of which are critical regulators of vital pathways. 25 Even the same HDAC may have different roles at different stages of the same disease. For example, HDAC1 was reported to be oncosuppressive in the early stage while oncogenic in established tumor cells in a mouse tumor model. 162 The inhibitors, even selective inhibitors of HDAC, could have many off-target effects. Undesired toxicity of certain HDACIs could lead to dysfunction of other important biological processes. For instance, selective Class I HDACIs showed potent anti-tumor effects, but inhibition of Class I HDACs, specifically HDAC3, caused impaired DNA damage response. 163 Other major concerns include the cardiac toxicity by some HDACIs, 164 and the cellular resistance to certain HDACIs. 165 A study in phase I and II clinical trials of romidepsin in patients with cutaneous T-cell lymphoma, de novo resistance of the drug was found. 165 The lack of response-predictive markers for cardiac and pulmonary fibrosis also delayed advances in establishing the valuable index for therapeutic efficacy. To overcome these limitations and side effects, a better designed, more selective, and better targeted HDACI would increase the potency against fibrotic diseases. For pulmonary fibrosis, localized administration of the drug (such as nasal delivery) would minimize the undesired whole-body side effects.
Conclusion
Studies in the past have shown that HDACs are critical in the pathogenesis of fibrosis by modulating acetylation of histone and nonhistone substrates. HDACs are found to be dysregulated in fibrotic diseases. Inhibition of HDACs in fibrotic models mitigates fibrotic status. HDACIs, especially selective inhibitors, would offer better understanding of how HDACs participate in the process of fibrogenesis. Inhibitors of HDACs would offer a novel category of therapeutic options in fibrosis. Extensive studies are needed to clarify the specific roles of each HDAC in the fibrotic process, which would form a mechanistic foundation for clinical usage. Many of the current HDACIs in clinical trials are broad-spectrum nonselective inhibitors and have more toxicity and unwanted side effects than selective HDACIs. Right now, most HDACIs either approved by the FDA or in clinical trials are mainly for cancer treatments. HDACIs for cardiac or pulmonary fibrosis are still mostly in preclinical stages. Better targeted, more specific HDACIs are needed to enhance the therapeutic efficacy and reduce toxicity. Furthermore, it is crucial to develop biomarkers for HDACI alone or in combination with other reagents to predict the response of individual patient to treatment. Elucidation and validation of the mechanisms of HDAC involved in the pathogenesis of fibrosis would provide a powerful tool in the treatment of fibrotic-related diseases, especially cardiac and pulmonary fibrosis.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the National Natural Science Foundation of China (grant number 81470256 to X.Z.) and US National Institutes of Health (grant number R01AG050567 to Y.Y.S.).
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.