
Abstract
The obesity pandemic presents a significant burden, both in terms of healthcare and economic outcomes, and current medical therapies are inadequate to deal with this challenge. Bariatric surgery is currently the only therapy available for obesity which results in long-term, sustained weight loss. The favourable effects of this surgery are thought, at least in part, to be mediated via the changes of gut hormones such as GLP-1, PYY, PP and oxyntomodulin seen following the procedure. These hormones have subsequently become attractive novel targets for the development of obesity therapies. Here, we review the development of these gut peptides as current and emerging therapies in the treatment of obesity.
Background
The current obesity pandemic is posing a major challenge to healthcare providers. According to World Health Organization (WHO) global statistics, in 2008 10% of men and 14% of women were classified as obese, with a further 35% of adults categorized as overweight [WHO, 2012] This problem is exaggerated in Western countries and by 2011 in England alone, the proportion of overweight and obesity had risen to 65% of men, and 58% of women [Health and Social Care Information Centre, 2013]. This growing obesity epidemic represents a huge burden in terms of both health and economic outcomes. In 2007, NHS costs directly attributable to overweight and obesity amounted to £4.2 billion [Swanton, 2008], and are projected to rise to £8.3 billion by 2025, reaching almost 12% of the total NHS budget [Foresight, 2007]. In addition, obesity is an important risk factor for a number of other medical conditions including type 2 diabetes, ischaemic heart disease, stroke and cancer, and carries an increased risk of both all-cause and cause-specific mortality [Ringbäck Weitoft et al. 2008; Whitlock et al. 2009; Zheng et al. 2011].
In the face of this rising wave of obesity, there is a need for effective medical treatment strategies. However, therapeutic options, particularly medical therapies, are limited. Lifestyle and dietary modification alone, whilst initially promising, do not provide a long-term treatment option for the majority of obese individuals. Orlistat is the only currently licensed medical treatment for obesity in the UK. It is a pancreatic lipase inhibitor which prevents fat absorption, but achieves only a modest weight loss of 2.9 kg compared with placebo [Rucker et al. 2007]. Therapies such as sibutramine and rimonabant, which were previously licensed for obesity have been withdrawn due to concerns regarding cardiovascular and psychiatric health respectively [Christensen et al. 2007; James et al. 2010].
Successful obesity treatment is currently limited to bariatric surgery. This is the only therapy to have demonstrated the ability to produce a sustained, long-term weight loss and deliver reductions in mortality and morbidity [Sjostrom et al. 2007]. The most common bariatric procedure is the Roux-en-Y gastric bypass (RYGB), accounting for up to 75% of all weight loss surgeries [Smith et al. 2008]. It is now well-established that following gastric bypass, there is an alteration in the secretion of various gut hormones associated with appetite and satiety as well as energy expenditure (EE) [Beckman et al. 2011; Le Roux et al. 2006, 2007; Pournaras et al. 2010]. These changes are thought to contribute to the superior weight loss and improvement in blood glucose observed after RYGB. These findings have led to the suggestion that gut hormones could be targets for novel obesity and diabetes therapies, and there has been much subsequent interest in these peptides.
Gut hormones
Peptide YY
Peptide YY (PYY) is a 36-amino-acid peptide secreted from entero-endocrine L cells of the distal gut in response to an oral nutrient load (Figure 1). PYY belongs to the ‘PP fold’ family of peptides which includes pancreatic polypeptide (PP), and neuropeptide Y (NPY). They are so named because their tertiary structure forms a hairpin-like U-shaped fold.
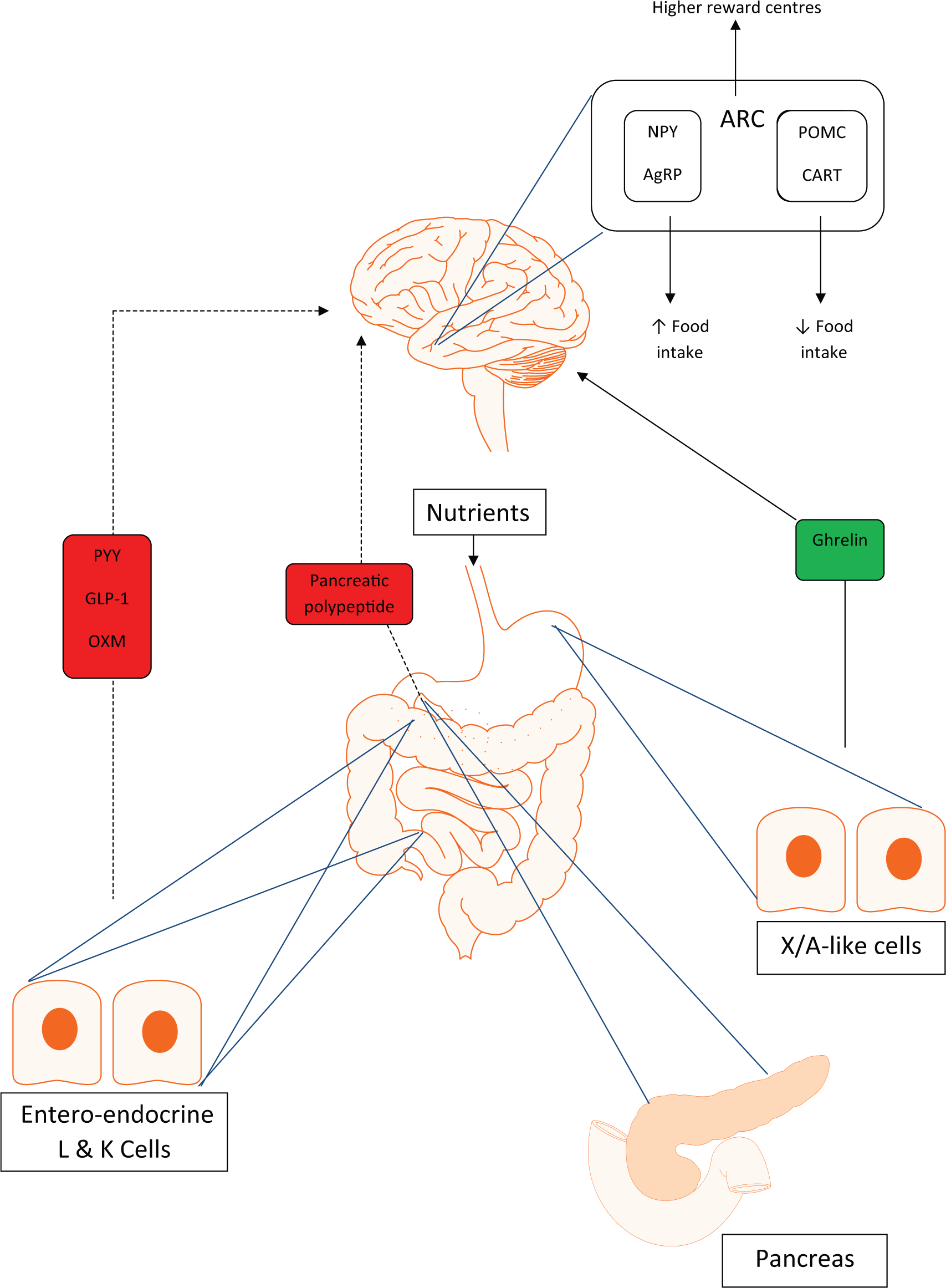
Interactions of the gut-brain axis. Solid line indicates positive feedback, and dashed line indicates negative feedback. Peptide YY (PYY), glucagon-like peptide-1 (GLP-1) and oxyntomodulin (OXM) all provide negative feedback to appetite-regulating areas within the hypothalamus on sensing an oral nutrient load. Pancreatic polypeptide (PP) from pancreatic islets is also anorectic, signalling via central appetite centres in the hypothalamus. Ghrelin is orexigenic, providing positive feedback to the hypothalamic nuclei, and activating neuropeptide Y (NPY) and agouti-related peptide (AgRP) neurones to increase food intake.
PYY secretion is stimulated by ingestion of all nutrients, however, opinions are varied as to which macronutrient causes the greatest stimulation of PYY release. In rodents, protein appears to provide the most potent stimulus for its release [Batterham et al. 2006], whereas a high-carbohydrate, low-fat diet was shown to give the highest levels in obese humans [Essah et al. 2007]. Interestingly, PYY levels start to rise within about 15 minutes of any caloric ingestion [Gibbons et al. 2013], long before the nutrients themselves reach the distal gut, implying other neural or hormonal mechanisms for its release. It is secreted as PYY1-36, and is then cleaved by the enzyme DPP-4 to give the active form, PYY3-36. PYY, along with other gut hormones such as oxyntomodulin (OXM) and glucagon-like peptide-1 (GLP-1), show augmented postprandial levels in subjects who have undergone gastric bypass surgery [Borg et al. 2006; Laferrère et al. 2010; Le Roux et al. 2006]. This rise in PYY is thought, at least in part, to be responsible for the marked and sustained weight loss seen in this patient group.
PYY acts via the Y2 receptor, a member of the NPY receptor family. Y2 receptors are found throughout both the peripheral and central nervous systems, with a greater concentration in the arcuate nucleus of the hypothalamus (ARC), an area known to be key in the regulation of appetite. The actions of PYY include a reduction in gastric emptying and a delay in intestinal transit, the so-called ‘ileal brake’. PYY has a potent inhibitory effect on food intake in both rodents and humans, as well as reducing body weight [Batterham et al. 2002]. PYY infusion has also been shown to reduce ghrelin levels, ghrelin being well characterized as an orexigenic hormone [Batterham et al. 2003b]. Basal levels of PYY are lower in obese subjects compared with their lean counterparts, and in obesity, there is also a blunted postprandial PYY rise [Batterham et al. 2003b], suggesting that a lack of endogenous PYY secretion may be implicated in the development of obesity. However, following infusion of PYY, obese and lean subjects alike experience a reduction in hunger and caloric intake [Batterham et al. 2003b], making it likely that exogenous PYY could be a successful therapy for obesity. In order to develop PYY as a treatment for obesity, there are obstacles to overcome. PYY has a very short half-life (t1/2) of just 7–10 minutes [Lluis et al. 1989], and a functional t1/2 of just 3 hours [Shechter et al. 2005], although Batterham and colleagues did show a sustained reduction in food intake over the subsequent 24 hours following a short, 90 minute infusion of PYY [Batterham et al. 2002]. Various approaches have been employed to extend the t1/2 of native PYY, and thus extend its functional capacity, one being to alter the amino acid sequence of the peptide to target cleavage sites and thus negate degradation. Long-acting analogues of PYY have been developed, to be administered subcutaneously. Phase I results looking at the effects of one of these analogues on safety, tolerability, food intake and body weight are awaited [ClinicalTrials.gov identifier: NCT01515319]. Other groups have tried PEGylation, which helps reduce renal clearance, and lessens the risk of immunogenicity. It has been previously observed that permanent PEGylation of PYY inactivates it, as the large PEG molecule potentially prevents adequate receptor binding for the smaller peptide [Shechter et al. 2005]. Shechter and colleagues developed a reversibly PEGylated PYY molecule, which achieved a longer functional t1/2, and gave an effect on satiety comparable to that achieved with PYY infusion [Shechter et al. 2005].
Peptide hormones do not lend themselves to oral administration due to degradation in the stomach, and injectable therapies are not acceptable to all patients, so some groups have developed alternative methods for PYY delivery. One phase II study used the nasal route to deliver PYY, the nasal mucosa being very vascular and thus allowing effective peptide absorption. This did indeed achieve PYY levels that were considered pharmacologically relevant, but at higher doses caused unacceptable nausea and vomiting due to sharp rises in plasma PYY levels [Gantz et al. 2007]. Oral PYY in mice was given via a spray delivering peptide to the buccal mucosa, and also through genetic manipulation to upregulate PYY expression in the salivary glands using a viral vector-mediated gene transfer. Both modalities appeared to reduce food intake and body weight, although salivary PYY was not detected in plasma [Acosta et al. 2011]. Oral administration of PYY was achieved in rats by conjugating PYY to vitamin B12 and using the B12 uptake pathway to facilitate peptide delivery [Fazen et al. 2011]. Emisphere has also used a delivery agent (sodium N-[8-(2-hydroxybenzoyl) amino]caprylate) to form a lipophilic complex that transports the peptide across membranes of the gastrointestinal tract [Steinert et al. 2010]. Steinert and colleagues saw promising results in phase I and II studies, with PYY alone achieving a 12% reduction in energy intake, albeit this was statistically nonsignificant. The overall key to achieving successful PYY therapy will lie in delivering stable levels for extended periods of time to avoid sharp peaks and subsequent development of nausea.
Pancreatic polypeptide
PP belongs to the same ‘PP fold’ family of peptides as PYY. Like PYY it is also a 36-amino-acid peptide produced after eating. However, whilst PYY comes from entero-endocrine L cells in the distal gut, PP is secreted by the PP cells of the islets of Langerhans in the pancreas (Figure 1) [Adrian et al. 1976; Larsson et al. 1975]. Similar to PYY, PP levels are found to be reduced in obese subjects, and exhibit a smaller rise following meals [Marco et al. 1980].
PP is known to reduce food intake in rodents [Asakawa et al. 2003b], in normal weight human subjects [Asakawa et al. 2006; Batterham et al. 2003a; Jesudason et al. 2007] and in Prader–Willi syndrome, a condition characterized by obesity and hyperphagia [Berntson et al. 1993]. PP has a high affinity for Y4 receptors, a subtype of the NPY receptor which is found widely in the brain including in the hypothalamus and brainstem, areas which are key in the central control of appetite [Parker and Herzog, 1999]. The importance of this central mechanism of PP action is supported by the fact that in mice with hypothalamic Y4 receptor deletion, PP was unable to induce c-fos activation (a marker of neuronal activation) [Lin et al. 2009].
In addition to reducing food intake, PP also reduces gastric emptying [Asakawa et al. 2003b; Schmidt et al. 2005], potentially providing feedback to central satiety centres via the vagus nerve. Vagal afferent signals are triggered on eating by gastric distension stretch receptors in the stomach [Wang et al. 2008]. Interestingly, Asakawa and colleagues, as well as demonstrating a reduction in gastric emptying, showed that vagotomy attenuated PP’s inhibitory effect on food intake. This provides evidence that at least some of its satiety actions are vagally mediated, maybe through gastric distension, but also potentially via PP receptors on the vagal afferents signalling to satiety centres in the brainstem and hypothalamus [Asakawa et al. 2003b]. The same group also postulate an increase in EE by PP. An increase in EE coupled with a reduction in food intake would augment any weight loss caused by administering PP to obese subjects [Asakawa et al. 2003b], making it an attractive option as an obesity therapy.
As mentioned previously, any potentially therapeutic peptide should offer acceptable routes of administration, and be compatible with daily (or less frequent) administration. Like PYY, PP has a short t1/2 of only 7 minutes [Adrian et al. 1978], although Batterham and colleagues demonstrated a prolonged effect on food intake after a 90-minute infusion of PP was stopped, with the reduction in energy intake lasting up to 24 hours post-infusion [Batterham et al. 2003a]. Thus, long-acting analogues of PP have been developed in order to overcome the short t1/2 and the need to deliver peptide as an infusion. Bloom and colleagues published their phase I study of a PP analogue, PP1420, last year [Tan et al. 2012]. They altered the amino acid sequence of PP adding a glycine residue at position 0, and substituting various other amino acids to make the analogue peptidase resistant. The t1/2 was increased to an average of 2.42–2.61 hours, with no tolerability issues. Further studies will need to take place to examine its effect on food intake. Another example of a PP analogue has been developed by Bellmann-Sickert and colleagues, which they describe as ‘lipidated’, or acylated with fatty acids [Bellmann-Sickert et al. 2011]. This addition of fatty acids allows the peptide molecule to bind to plasma proteins, providing stability and protection from proteolytic enzymes [Bellmann-Sickert et al. 2011]. They also assessed the effects of PEGylation of PP, examining both pharmacokinetics, and food intake in rodents. Whilst both molecules achieved a favourable effect in terms of pharmacokinetic profile, the lipidated peptide had a better resistance to degradation and clearance. Interestingly, the lipidated molecule also reduced food intake in mice, and to a greater extent than the PEGylated peptide. An additional challenge seen with PP is that it tends to form aggregates in aqueous solution. Adding side chains as previously discussed will to some extent negate this problem, but an alternative approach has been to deliver PP within micelles, which are themselves formed from PEGylated phospholipids. In vitro, the micellar formulation increased stability of PP by about 2.5-fold, whilst maintaining bioactivity as measured by cyclic AMP (cAMP) inhibition [Banerjee and Onyuksel, 2012]. The next step will be to investigate whether this preparation maintains its favourable properties in vivo, without adverse events.
The only clinical trial of a PP-based therapy is therefore that conducted by Bloom and colleagues mentioned above [Tan et al. 2012]. Any other data on the use of PP in humans has been generated using the native peptide with its inherent short t1/2. Further in vivo studies using modified forms of PP are necessary to evaluate its full potential as an anti-obesity agent.
Glucagon-like peptide-1
GLP-1 is a peptide hormone which acts as an incretin, stimulating release of insulin from the pancreatic β cells in response to an oral nutrient load. GLP-1 is released mainly from the entero-endocrine L cells in the small intestine (Figure 1) [Herrmann et al. 1995]. It is a product of post-translational processing of the glucagon precursor, proglucagon [Orskov et al. 1989].
GLP-1 has two major biologically active forms, GLP-1(7-36) amide and GLP-1(7-37), the predominant one being GLP-1(7-36) amide. GLP-1 is rapidly broken down in vivo by the enzyme dipeptidyl peptidase-4 (DPP-4) to inactive metabolites, and therefore has a short circulating t1/2, rendering native GLP-1 inappropriate for therapeutic use. GLP-1 acts via its own distinct receptor, a G protein coupled receptor, which has a varied tissue distribution in humans. Amongst others, GLP-1 receptors are found in the pancreas, gut and brain, in particular the hypothalamus, NTS, and area postrema (AP), all areas closely related to appetite regulation [Merchenthaler et al. 1999]. In addition to its incretin effect, actions of GLP-1 include suppression of glucagon secretion from α cells of the pancreas [Creutzfeldt et al. 1996], delaying gastric emptying [Schirra et al. 2006], and suppression of appetite [Punjabi et al. 2011].
GLP-1 is currently the most successful gut hormone to be exploited for therapeutic purposes in humans. It is used for treatment of type 2 diabetes due to its incretin effect, and to date several preparations are available. The first molecule to be developed as a drug was exenatide (Byetta®). It was first approved by the US Food and Drug Administration (FDA) in 2005, and was derived from the saliva of the Gila Monster lizard Heloderma suspectum. It shares only 53% homology with human GLP-1. Whilst it binds to the GLP-1 receptor, it is resistant to degradation by DPP-4, lengthening the t1/2 and allowing twice-daily injection [Robles and Singh-Franco, 2009]. Following this, other manipulations of GLP-1 were made to extend the t1/2, and provide a more acceptable injection schedule. Liraglutide (Victoza®) was developed as a once-a-day injection alternative to exenatide. Liraglutide has a much closer homology (97%) to human GLP-1, but has been modified by the addition of a 16-carbon fatty acid chain which allows it to bind to albumin [Sisson, 2011]. This negates breakdown and clearance and allows the more desirable once daily administration. In a head-to-head study, liraglutide performed better than exenatide in terms of glycaemic improvement, but the degree of body weight loss was similar in both groups, and modest at ~3 kg in 26 weeks [Buse et al. 2009]. Nausea is the main side effect of these GLP-1 receptor agonists, and prevents the administration of higher doses which might have greater effects on weight loss. Further progress has been made with the advent of once-weekly exenatide LAR or Bydureon®. Exenatide is incorporated in microspheres which consist of poly(D,L lactic-co-glycolic acid), which allows for a gradual delivery of the drug through slow-release technology [Drucker et al. 2008 ]. In addition to good effects on glycaemic control, the overall amount of weight loss over a 30-week study was similar to that achieved with exenatide itself [Drucker et al. 2008]. Since the success of these GLP-1 receptor agonists in the treatment of diabetes, many others are in the pipeline, with prolonged t1/2 due to variations in molecular structure. However, despite encouraging results for diabetes control, the utility of these as weight loss therapies remains limited and it seems unlikely that GLP-1 monotherapy will be licensed as a treatment for obesity. Phase III trials are currently underway to assess an osmotic minipump, which provides a constant subcutaneous delivery of exenatide [Tibble et al. 2013] (see also http://www.intarcia.com/products-technology), hopefully avoiding the peak peptide levels more likely to cause the commonest side effect, nausea. Phase II studies of this technology reported encouraging weight loss, and so results of the phase III study will be revealing.
Recently, concerns have been raised over the safety of GLP-1 receptor agonists. It is suggested that both DPP-4 inhibitors (which prevent breakdown of GLP-1), and GLP-1 receptor agonists increase the potential risk of pancreatitis, pancreatic cancer, hyperplasia of the exocrine pancreas, and thyroid C-cell hyperplasia, potentially a precursor of medullary thyroid cancer [Butler et al. 2013]. However, these findings remain under investigation, and no conclusion with regards to causality has yet been drawn. The risk-benefit ratio of the use of these agents must also be considered [Nauck, 2013].
Oxyntomodulin
OXM, a 37-amino-acid peptide hormone is processed similarly to GLP-1, from post-translational processing of proglucagon. It is cosecreted with GLP-1 from the entero-endocrine L cells in response to a meal (Figure 1). OXM binds to both the GLP-1 and glucagon receptors but it seems likely that it exerts the majority of its effect on appetite via the GLP-1 receptor, as co-administration of a GLP-1 receptor antagonist blocks the anorectic actions of OXM [Dakin et al. 2001].
OXM has been shown to inhibit both gastric acid secretion and pancreatic enzyme secretion, in addition to delaying gastric emptying following intravenous administration [Schjoldager et al. 1989]. Given peripherally in rats, OXM has been demonstrated to significantly decrease food intake, as well as reducing body weight gain compared with pair fed animals [Dakin et al. 2004]. As pair fed and treatment groups receive identical amounts of food, the group treated with OXM must have a further stimulus to weight loss apart from decreased food intake. On examination of the fat pads of both groups of animals, the fat pad weight was lower in OXM-treated rats. This suggested an increase in EE to be the added stimulus to weight loss, with the OXM group utilizing fat stores as an energy substrate. OXM has been shown to increase EE [Dakin et al. 2002; Wynne et al. 2006] which appears to be mediated via an increase in activity related EE. We recently showed that exogenously administered glucagon reliably increases resting EE [Tan et al. 2013] As OXM has dual actions at both the glucagon and GLP-1 receptors, it is likely that it exerts its effects on EE via the glucagon receptor.
When injected centrally into the ARC, OXM causes a sustained inhibition of food intake [Dakin et al. 2004]. Intravenous infusion of OXM in healthy human volunteers has been reported to decrease food intake [Cohen et al. 2003] both during, and in the 12 hours following the end of the infusion. Furthermore, subcutaneous self-administration of OXM over a 4-week period gave a 2.4% reduction in body weight, and reduced food intake in overweight and obese volunteers [Wynne et al. 2005].
OXM presents an exciting prospect for the treatment of obesity due to its dual effects on both food intake reduction, and increase in EE: a two-pronged attack on obesity. It appears to be able to act at the glucagon receptor to increase EE with none of the unfavourable effects on glycaemia that might be expected from glucagon receptor agonism. A combination approach, with action at both the glucagon and GLP-1 receptors has also been proven to be successful in rodent studies which employed a peptide analogue dual agonist of these two systems. These studies revealed weight loss with no deterioration of glycaemic control, and an improved lipid profile in diet-induced obese animals [Day et al. 2009; Pocai et al. 2009], which provides an encouraging avenue for future development of obesity therapies. The ratio of agonism between the GLP-1 and glucagon receptors will be crucial to achieve a maximum reduction in food intake, with optimal effects on EE and glycaemic control. Zealand Pharma is developing a similar dual agonist, ZP-2929, which improves glycaemic control without causing weight gain [Fosgerau et al. 2011]. Thiakis/Pfizer has also developed analogues which are dual agonists at the GLP-1 and glucagon receptors, and at least one of these, TKS1225/OAP-189 has undergone phase I/II trials for safety and tolerability.
Ghrelin
Ghrelin is the only known orexigenic hormone, and when administered to human volunteers, both lean and obese, it increases food intake [Druce et al. 2005, 2006]. It is a 28-amino-acid peptide secreted from X/A-like cells in the fundus of the stomach (Figure 1) and is highest in the fasted state, with levels falling postprandially. Ghrelin exists in two main forms, the inactive, nonacylated form, and the active, acylated form, converted from nonacylated ghrelin by the enzyme Ghrelin O-acyltransferase (GOAT). Like PP, PYY and other gut hormones, ghrelin has receptors within the hypothalamus which may contribute to its effect on appetite [Mondal et al. 2005; Willesen et al. 1999]. Acyl-ghrelin, the active form, is known to decrease insulin secretion [Tong et al. 2013]
Interestingly, ghrelin suppression after a meal was not related to vagal signalling, but the elevation of ghrelin seen in the fasted state was completely abolished by subdiaphragmatic vagotomy [Williams et al. 2003], suggesting that appetite suppression, in addition to being mediated through the hypothalamus, may also be controlled via other pathways.
Levels of ghrelin rise in people with dietary weight loss or anorectic disorders [Cummings et al. 2002; Shiiya et al. 2002], but in contrast are low in obesity, and after gastric bypass surgery [Cummings et al. 2002; Shiiya et al. 2002]. If ghrelin levels could be lowered or the receptor antagonized, then this could potentially be exploited as an obesity therapy. Ghrelin antagonists and receptor blockers are therefore a group of molecules which have received some interest for development as anti-obesity targets [Schellekens et al. 2010]. Asakawa and colleagues demonstrated a potent effect on food intake reduction with both peripheral and central administration of ghrelin receptor antagonists [Asakawa et al. 2003a]. Several other molecules have been developed, some with potential, but some with very limited in vivo action [Schellekens et al. 2010]. Also of interest, Shearman and colleagues used an RNA ‘Spiegelmer’, a synthetic polyethylene glycol (PEG)-modified L-RNA oligonucleotide, to bind acyl, or active, ghrelin and neutralize its in vivo effect. By neutralizing active ghrelin, they demonstrated a reduction in food intake, and body weight loss in mice [Shearman et al. 2006]. Knockout of the GOAT enzyme that activates ghrelin in vivo has been demonstrated to reduce hedonic feeding behaviour in mice [Davis et al. 2012], and therefore opened up a further possible avenue for ghrelin therapeutics. Other groups have subsequently manipulated GOAT, demonstrating end-product inhibition of GOAT activity [Yang et al. 2008], and it remains a potential target for obesity treatment [Gualillo et al. 2008]. A further therapeutic approach has been the development of a ‘vaccine’ against ghrelin, by injecting rats with ghrelin immunoconjugates. Results were promising, with a reduction in body weight gain and preferential decline of body fat [Zorrilla et al. 2006].
Combination therapies
Despite promising results with some of these gut hormones as monotherapies, if we truly wish to replicate the gut hormone changes seen after bariatric surgery, then better results may be obtained by combining the effects of more than one hormone. In 2005, Neary and colleagues showed that PYY and GLP-1 have an additive effect on food intake both when given peripherally in mice, and following intravenous infusion in man [Neary et al. 2005]. The same group subsequently investigated the combination of PP and PYY, however there was no benefit in the combination of these two hormones over either peptide alone when given in mice or humans [Neary et al. 2008]. The combination of PYY and OXM acts on NPY receptors, as well as both the GLP-1 and glucagon receptors, and this combination also shows a reduction in food intake compared with either hormone alone [Field et al. 2010]. In addition, we must not forget the dual agonism effects of GLP-1 and glucagon described above [Day et al. 2009; Pocai et al. 2009]. The use of dual or even triple agonists of the hormones described above may hold the key to maximizing body weight loss and developing an optimal obesity treatment.
Conclusion
In order to tackle the global obesity problem, new treatment options need to be developed outside of the operating theatre. They must promote a shift in the balance between energy intake and expenditure by increasing satiety, decreasing appetite, and encouraging an increase in energy output. The properties of gut hormones, and their known effects on EE and intake fulfil these criteria and make them prime targets for the development of new obesity therapies.
Footnotes
Funding
The Section is funded by grants from the MRC, BBSRC, NIHR, an Integrative Mammalian Biology (IMB) Capacity Building Award, an FP7- HEALTH- 2009- 241592 EuroCHIP grant and is supported by the NIHR Imperial Biomedical Research Centre Funding Scheme. R.C.T. is supported by a Clinical Research Fellowship from the MRC. T.M.T. is supported by grants from the MRC. S.R.B. is supported by an NIHR Senior Investigator Award and the MRC.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.