
Abstract
Type 2 diabetes (T2D) is a highly prevalent disorder that affects millions of people worldwide. The hallmark of T2D is hyperglycemia and, while many treatment modalities exist, achieving and maintaining glycemic control can be challenging. Glucagon-like peptide-1 (GLP-1) receptor agonists (RAs) are an appealing treatment option as they improve glycemic control, reduce weight, and limit the risk of hypoglycemia. Lixisenatide is a once-daily GLP-1 RA that has been evaluated in the GetGoal clinical trial program and has demonstrated efficacy and tolerability across a spectrum of patients. The feature that most distinguishes lixisenatide from other GLP-1 RAs is its ability to substantially reduce postprandial glucose (PPG) for the meal immediately following injection. Because of its positive effects on PPG, lixisenatide is being considered as a replacement for prandial insulin, and a fixed dose combination product containing lixisenatide and basal insulin is in development. Lixisenatide is a promising new addition to the antidiabetic armamentarium, but due to the lack of real-world experience with the drug, its exact place in therapy is unknown.
Introduction
By the year 2030 it is estimated that more than 360 million people worldwide will be afflicted with type 2 diabetes (T2D) [Wild et al. 2004]. T2D is a progressive disease characterized by worsening glycemic control, which underscores the need for effective pharmacotherapy options for managing hyperglycemia. A significant therapeutic advancement in the management of hyperglycemia is the advent of glucagon-like peptide-1 (GLP-1) receptor agonists (RAs).
GLP-1 RAs activate the GLP-1 receptor to increase glucose-dependent insulin secretion and satiety, decrease inappropriate glucose-dependent glucagon secretion and slow gastric emptying [Inzucchi et al. 2012]. Lixisenatide is one such agent developed by Sanofi and marketed by Zealand Pharma A/S (Copenhagen, Denmark) as Lyxumia® in Europe; approval of lixisenatide in the US is expected in late 2015. Lixisenatide is a once-daily prandially-acting GLP-1 RA for the treatment of T2D. The ability of lixisenatide to reduce glycosylated hemoglobin (A1C), fasting plasma glucose (FPG) and body weight is similar to or less than other GLP-1 RAs and dipeptidyl-peptidase 4 (DPP-4) inhibitors; however, lixisenatide has a pronounced ability to decrease postprandial glucose (PPG). This feature distinguishes it from the longer acting GLP-1 RAs and makes it an ideal agent for patients who experience PPG excursions. This review discusses the pharmacology, efficacy, safety and place in therapy of lixisenatide.
Pharmacology
Mechanism of action and dosing
Lixisenatide is a synthetic analog of endogenous exendin-4 that acts as a selective GLP-1 RA. Compared with exendin-4, lixisenatide contains a C-terminal modification of the addition of six lysine residues and deletion of a proline that increases its binding affinity to the GLP-1 receptor and increases its circulating halflife (t1/2) [Werner et al. 2010]. Lixisenatide has a four-fold higher binding affinity of the GLP-1 receptor compared with native GLP-1 [Liu et al. 2010]. Like other GLP-1 RAs, lixisenatide suppresses inappropriate glucagon secretion from pancreatic α cells, stimulates glucose-dependent insulin secretion by pancreatic βcells and increases feelings of satiety by delaying gastric emptying [Holst et al. 2008].
The starting dose of lixisenatide is 10 μg subcutaneously once daily for 14 days followed by 20 μg once daily thereafter. It is recommended that lixisenatide be administered within 1 hour of the same meal each day [Sanofi, 2014]. Clinical trials have evaluated administration of lixisenatide prior to a standardized breakfast [Kapitza et al. 2013; Seino et al. 2014; Meier et al. 2015; Ahren et al. 2013; Yu Pan et al. 2014; Rosenstock et al. 2015].
Pharmacokinetics
Lixisenatide pharmacokinetics (PK) were established during phase I and II clinical trials. Following a 20 μg dose of lixisenatide, drug concentrations peaked in 1–2 hours and the drug had a t1/2 of 2–4 hours [Distiller and Ruus, 2008; Becker et al 2010]. Despite the relatively short t1/2, lixisenatide is effective when dosed once daily. Explanations of this phenomenon include the high affinity of lixisenatide for the GLP-1 receptor and inhibition of gastric emptying [Petersen and Christensen, 2013]. Lixisenatide 20 μg once-daily dosing results in a mean Cmax of 187.2 pg/ml [Distiller and Ruus, 2008]. A phase IIa dose-ranging study demonstrated a correlation between increasing doses and increasing area-under-the-curve (AUC) concentrations of lixisenatide [Distiller and Ruus, 2008].
In a small phase I study, 24 patients with severe renal impairment (CrCl < 30 ml/min, not requiring dialysis) experienced a significant increase in lixisenatide AUC (346 ± 116 h·pg/ml) compared with patients with normal renal function (CrCl > 80 ml/min; AUC = 210 ± 90 h·pg/ml) following a single 5 μg dose [Liu and Ruus, 2009]. However, the prescribing information for lixisenatide does not recommend a dose adjustment based on renal impairment. Limited experience in this population warrants caution when lixisenatide is utilized in patients with CrCl 30–50 ml/min and manufacturer recommendations include avoiding use in patients with CrCl < 30 ml/min [Sanofi, 2014]. To date there are no published studies evaluating the PK of lixisenatide in patients with hepatic impairment.
A small phase I study of elderly patients demonstrated that lixisenatide is safe and effective in an older population. Raccah and colleagues evaluated lixisenatide PK in 18 healthy elderly (age ⩾65 years) subjects matched with 18 healthy younger (age 18–45 years) subjects. Subjects were given a one-time dose of lixisenatide 20 μg. Upon analysis it was found that the elderly subjects were exposed to approximately 30% more lixisenatide compared with their younger counterparts [AUC 1.29; 90% confidence interval (CI): 1.06–1.57] and the t1/2 was prolonged by about 1.6 times in this population as well. The Cmax and Tmax were similar between groups [Raccah et al. 2015]. Despite the increased drug exposure demonstrated in this small study, there are no age-specific dosing recommendations in the prescribing information for lixisenatide [Sanofi, 2014].
Pharmacodynamics
Lixisenatide pharmacodynamics (PD) have been established in patients with uncontrolled T2D receiving metformin, sulfonylureas or both. A study by Ratner and colleagues evaluated the dose-response in patients inadequately controlled on metformin who received lixisenatide 5, 10, 20 or 30 μg once daily or twice daily (BID) versus placebo. The mean baseline A1C was 7.55%. Patients receiving lixisenatide 5, 10, 20 or 30 μg once daily had least square (LS) mean reductions in A1C of 0.47%, 0.50%, 0.69% and 0.76%, respectively, and patients receiving lixisenatide 5, 10, 20 or 30 μg BID had LS mean reductions in A1C of 0.65%, 0.78%, 0.75% and 0.87%, respectively (all p < 0.01 versus placebo, which had an A1C reduction of 0.18%). Reductions in FPG ranged from −0.19 mmol/l (lixisenatide 5 μg BID) to −1.42 mmol/l (lixisenatide 30 μg BID) compared with −0.21 mmol/l for placebo (p < 0.01 for lixisenatide 30 μg daily, 10 μg BID, 20 μg BID and 30 μg BID versus placebo). Lixisenatide significantly decreased 2-hour PPG compared with placebo (range −2.01 to −4.61 mmol/l for lixisenatide versus −0.41 mmol/l for placebo; p < 0.05 for all lixisenatide doses). The percentage of patients who discontinued treatment due to adverse effects (AEs) ranged from 1.8% to 14.8% for lixisenatide compared with 1.8% for placebo. More AEs were noted in patients receiving lixisenatide BID and for patients receiving more than 20 μg daily. The results of this study demonstrated a dose–response relationship with lixisenatide and established the lixisenatide 20 μg once daily as the preferred dose to use in phase III clinical trials [Ratner et al. 2010].
Kapitza and colleagues assessed the PD parameters of once-daily lixisenatide compared with once-daily liraglutide in German patients with uncontrolled T2D receiving a stable dose of ⩾1500 mg of metformin. In addition to their background metformin dose, patients were randomized to receive either lixisenatide 10 μg once daily for 2 weeks followed by 20 μg once daily for 2 weeks or liraglutide 0.6 mg subcutaneously once daily for 1 week, then 1.2 mg once daily for 1 week, and then 1.8 mg once daily for 2 weeks. Glucose parameters were assessed using a standardized solid breakfast control. At 28 weeks, lixisenatide showed superior PPG reduction (−12.6 versus −4.0 h·mmol/l; p < 0.001) as well as significantly greater reductions in PPG excursions (−3.9 versus −1.4 mmol/l; p < 0.001) compared with liraglutide. These postprandial effects of lixisenatide led to significant reductions in postprandial insulin, C-peptide and glucagon concentrations compared with liraglutide (all p < 0.05). Liraglutide significantly reduced FPG and A1C compared with lixisenatide (−1.3 versus −0.3 mmol/l, p < 0.001 and −0.51% versus −0.32%, p < 0.01, respectively) [Kapitza et al. 2013].
Seino and colleagues evaluated the PD of lixisenatide in Caucasian and Japanese patients with T2D poorly controlled on stable doses of sulfonylureas with or without metformin. The primary endpoint of the study was the change in baseline PPG AUC after a standardized breakfast. Patients were continued on their background doses of sulfonylurea and/or metformin and were randomized to receive placebo or increasing doses of lixisenatide. Patients in the lixisenatide group started with either 5 μg once daily or BID or 10 μg once daily or BID, and were titrated up in 5 μg increments weekly to a total of 30 μg daily in the once daily groups and 60 μg daily in the BID groups. After 5–6 weeks of therapy, the LS mean differences in PPG AUC were −333.4 and −288.8 h·mg/dl for lixisenatide once daily and BID, respectively, versus placebo (p < 0.0001 for both). When reported by ethnic group, the LS mean difference in PPG AUC in Japanese patients was −406.7 and −346.3 (35.1) h·mg/dl for lixisenatide once daily and BID, respectively, versus placebo (p < 0.0001 for both) and in Caucasian patients was −260.1 and −231.3 h·mg/dl for lixisenatide once daily and BID, respectively, versus placebo (p < 0.0001 for both). When the LS mean difference in PPG AUC data were pooled versus placebo, the difference between the Japanese and Caucasian patient populations was −122.3 h·mg/dl (95% CI −211.10 to −33.51; p = 0.0074), showing a more pronounced PPG reduction in the Japanese population. In both Japanese and Caucasian patients, a maximal PPG AUC was achieved with the lixisenatide 20 μg daily dose. Across all populations, the once-daily lixisenatide dose produced greater reductions in PPG compared with the BID dosing. BID lixisenatide dosing was more effective at reducing FPG and hemoglobin A1C (A1C) [Seino et al. 2014].
A phase IIb study was conducted between lixisenatide and liraglutide to evaluate the PD and safety of the two agents being compared. A total of 142 patients with T2D optimized on insulin glargine with or without metformin were randomized in open-label fashion to receive either lixisenatide 20 μg, liraglutide 1.2 mg or liraglutide 1.8 mg once daily for 8 weeks. The primary endpoint was change in PPG AUC following a standardized meal test. Other efficacy parameters included change in gastric emptying, 24-hour plasma glucose profile, A1C, FPG, 24-hour ambulatory heart rate (HR) and blood pressure, and amylase and lipase levels. At the end of 8 weeks, patients receiving lixisenatide had significant reductions in PPG AUC (−108.3 h·mg/dl versus liraglutide 1.2 mg and −83.0 h·mg/dl versus liraglutide 1.8 mg; p < 0.001 for both). Reductions in 24-hour glucose profiles were similar between groups. Lixisenatide showed the most change in glucose postbreakfast, whereas liraglutide demonstrated sustained glucose reductions throughout the day. Although it was only an 8 week study, changes in A1C were reported. Liraglutide reduced A1C slightly more than lixisenatide (−0.1% for liraglutide 1.2 mg and −0.2% for liraglutide 1.8 mg; p = 0.17 and p = 0.007, respectively). Although the difference in A1C change between liraglutide 1.8 mg and lixisenatide was statistically different at 8 weeks, the clinical difference is negligible. Had the study duration been longer, the difference in A1C reduction may have become clinically meaningful. All patients experienced reductions in FPG and body weight that were not significantly different between groups. There were significant gastric emptying delays demonstrated with lixisenatide compared with liraglutide, which may explain the substantial decrease in PPG AUC and postbreakfast effect shown in the 24-hour glucose profiles. Both lixisenatide and liraglutide are dosed once daily, but have some distinct PD differences that may lead to patient-specific preferences [Meier et al. 2015].
The lixisenatide PK and PD data established the recommended dose and frequency moving forward into phase III clinical trials.
Efficacy: summary of phase III studies
Lixisenatide efficacy has been comprehensively evaluated through the GetGoal clinical trial program. The GetGoal program included a diverse patient population from Europe, Asia and America and several studies focused specifically on Asian patients with T2D (GetGoal-Mono-Japan, GetGoal-L-Asia and GetGoal-M-Asia). Lixisenatide has been studied against placebo as monotherapy and as adjunctive therapy to both oral antidiabetics (OADs) and insulin. It has been compared head-to-head with BID exenatide as well as daily and thrice daily insulin glulisine. The comprehensive phase III program has established the efficacy of lixisenatide in a variety of patient populations and combinations of antidiabetic therapies; the data are summarized in Table 1.
Phase III clinical trials of lixisenatide: efficacy data.

A1C, glycosylated hemoglobin; NR, not reported; PPG, postprandial glucose; TZD, thiazolidinedione.
Placebo comparators: use as monotherapy
Lixisenatide was evaluated as monotherapy in patients with T2D in the 12-week randomized, double-blind, placebo-controlled trial GetGoal-Mono. Patients not receiving any glucose-lowering therapy who had an A1C of 7–10% received lixisenatide or placebo injections in either a one- or two-step regimen: lixisenatide 10 μg daily for 1 week, then 15 μg daily for 1 week, and then 20 μg daily thereafter (2-step); lixisenatide 10 μg daily for 2 weeks, then 20 μg daily thereafter (1-step); placebo (2-step); or placebo (1-step). The two placebo arms were ultimately combined for data analysis purposes. The primary efficacy endpoint was change in A1C from baseline to 12 weeks. Secondary efficacy endpoints included percentage of patients achieving an A1C < 7% or ⩽6.5%, change in 2-hour PPG and 2-hour glucose excursions following a standardized meal test, and change in FPG between baseline and 12 weeks.
A total of 361 patients were enrolled and evaluated. Lixisenatide significantly reduced A1C from baseline after 12 weeks of therapy. The LS mean change in A1C from baseline was −0.19%, −0.73% and −0.85% for the combined placebo groups, 2-step and 1-step dose increase groups, respectively. The changes in A1C versus placebo were −0.54% and −0.66% for the 2-step and 1-step dose increase groups, respectively (both p < 0.0001). Significant percentages of patients in the lixisenatide groups achieved A1C values of <7% [52% (2-step) and 47% (1-step) versus 27% placebo; p < 0.001 for both] and ⩽6.5% [32% (2-step) and 25% (1-step) versus 13% for placebo; p < 0.01 for both] compared with placebo. More than 55% of patients in each lixisenatide arm developed anti-lixisenatide antibodies; however, the A1C reduction was similar whether antibodies were present or absent. In the 169 patients who participated in the standardized meal evaluation, lixisenatide significantly reduced both the 2-hour glucose excursion [LS mean change versus placebo of −3.1 mmol/l (2-step) and −3.7 mmol/l (1-step); p < 0.0001 for each] and 2-hour PPG [LS mean change versus placebo of −3.86 mmol/l (2-step) and −4.82 mmol/l (1-step); p < 0.0001 for each]. Lastly, the decrease in FPG was also significant when comparing lixisenatide with placebo [LS mean change versus placebo of −0.9 mmol/l (2-step) and −1.1 mmol/l (1-step); p < 0.001 and p < 0.0001, respectively]. Decreases in body weight were similar and not statistically different between groups. The authors concluded that lixisenatide monotherapy was effective in the treatment of T2D with a pronounced postprandial effect [Fonseca et al. 2012].
A small open-label study of lixisenatide monotherapy in 69 Japanese patients with T2D also demonstrated efficacy. The patients were randomized in open-label fashion to the lixisenatide 1- or 2-step dose titration algorithm as in the GetGoal-Mono. At 24 weeks, patients in each arm experienced a meaningful decrease in A1C [−0.74% (1-step) and −0.99% (2-step)] that was sustained at 76 weeks in the combined cohort (−0.72%). Patients in each arm also lost weight [1.08 kg (1-step) and −0.43 kg (2-step)] and experienced reduced FPG concentrations [−0.56 mmol/l (1-step) and −1.16 mmol/l (2-step)], both of which were sustained at 76 weeks (−1.58 kg and −0.46 mmol/l, combined group data) [Seino et al. 2012a]. Interestingly, the glycemic efficacy was more marked in the 2-step titration group in this study compared with the results in the Fonseca study [Fonseca et al. 2012]. As in the GetGoal-Mono study, lixisenatide was shown to be an efficacious monotherapy option in patients with T2D.
Placebo comparators: use in patients receiving oral antidiabetics
The GetGoal-F1, GetGoal-M and GetGoal-M-Asia studies all evaluated lixisenatide use compared with placebo in patients with T2D insufficiently controlled on metformin. The GetGoal-F1 study evaluated 484 patients receiving >1500 mg of metformin daily with an A1C of 7–10%. Patients were randomized to lixisenatide 1-step dose titration (10 μg once daily for 2 weeks followed by 20 μg once daily), lixisenatide 2-step dose titration (10 μg once daily for 1 week, then 15 μg once daily for 1 week, then 20 μg once daily), placebo 1-step dose titration, or placebo 2-step dose titration. The primary efficacy endpoint was change in A1C at 24 weeks. Secondary efficacy endpoints included the percentage of patients achieving an A1C < 7%, FPG and change in body weight. Both titration schemes of lixisenatide significantly reduced A1C from baseline compared with placebo (LS mean change of −0.5% for 1-step titration and −0.4% for 2-step titration; p < 0.0001 for both) and more patients receiving lixisenatide achieved A1C values of <7% and ⩽6.5% compared with placebo (p < 0.001 for both). The LS mean difference in FPG for lixisenatide compared with placebo was −0.7 mmol/l for both titration schemes (p < 0.001 versus placebo). Finally, patients in the lixisenatide groups lost more weight than those receiving placebo; the LS mean difference change compared with placebo was −1.0 and −1.1 kg for lixisenatide 1-step and 2-step groups, respectively (p < 0.01) [Bolli et al. 2014].
Similarly, GetGoal-M and GetGoal-M-Asia evaluated the efficacy of lixisenatide in patients with T2D insufficiently controlled on metformin. The GetGoal-M study included 680 patients with T2D who were receiving at least 1500 mg of metformin daily with an A1C of 7–10%. In the GetGoal-M-Asia study, patients could also be on a sulfonylurea. The patients in GetGoal-M and GetGoal-M-Asia were randomized to lixisenatide 20 μg once daily or placebo. The GetGoal-M study further stratified patients to morning or evening dosing of either drug or placebo. The primary efficacy endpoint was change in A1C at 24 weeks for both studies. Patients in the GetGoal-M study had a significant A1C decrease compared with placebo (LS mean difference of −0.5% for the morning dose cohort, −0.4% for the evening dose cohort; p < 0.0001 for each). Patients in the GetGoal-M-Asia had a similar reduction in A1C versus placebo (LS mean difference of −0.36%, p = 0.0004). All patients receiving lixisenatide across both studies were more likely to achieve an A1C < 7% or ⩽6.5% compared with those receiving placebo. Both studies also showed a reduction in 2-hour PPG following a standardized breakfast for lixisenatide compared with placebo [LS mean difference of −4.5 mmol/l (GetGoal-M morning dose cohort0, −4.28 mmol/l (GetGoal-M-Asia); p < 0.0001 for each]. Lixisenatide was also effective at reducing FPG compared with placebo, though to a lesser degree than PPG [LS mean difference −0.9 mmol/l (GetGoal-M morning dose cohort; p < 0.0001), −0.6 mmol/l (GetGoal-M evening dose cohort; p = 0.0046), −0.48 mmol/l (GetGoal-M-Asia; p = 0.0109)]. There were no significant differences in weight loss compared with placebo as patients in all groups lost weight. Both studies concluded that lixisenatide 20 μg once daily was effective at improving A1C and especially effective at reducing PPG [Ahren et al. 2013; Yu Pan et al. 2014].
Comparable A1C and PPG improvements were demonstrated in the GetGoal-S study that included patients with T2D who were inadequately controlled on OADs. A total of 859 eligible patients were randomized to receive either lixisenatide 20 μg once daily or placebo. All patients were receiving a sulfonylurea as background therapy and 84% were concurrently receiving metformin. Approximately half of the patients were Caucasian and slightly less than half were Asian. The primary efficacy endpoint was change in A1C at 24 weeks and secondary efficacy endpoints included percentage of patients achieving A1C values <7% and ⩽6.5%, changes in FPG, 2-hour PPG and body weight. At 24 weeks, the LS mean difference in A1C between lixisenatide and placebo was −0.84% (p < 0.0001). Significantly more patients receiving lixisenatide achieved one or both goal A1C values compared with placebo (36.4% versus 13.5% for A1C < 7% and 19.3% versus 4.7% for A1C ⩽ 6.5%; p < 0.001 for each). Patients receiving lixisenatide experienced statistically significant decreases in FPG (LS mean difference −0.63 mmol/l, p < 0.0001) and body weight (LS mean difference −0.84 kg, p < 0.0001) and a marked decrease in 2-hour PPG following a standardized meal test (LS mean difference −5.98 mmol/l, p < 0.0001) compared with placebo. The study authors concluded that lixisenatide was effective at reducing A1C from baseline with a pronounced postprandial effect [Rosenstock et al. 2014].
Lastly, a study by Pinget and colleagues evaluated the addition of lixisenatide to pioglitazone with or without metformin. Patients receiving a stable dose of ⩾30 mg per day of pioglitazone with or without a stable dose of ⩾1500 mg per day of metformin were randomized to receive either lixisenatide 20 μg once daily or placebo. The primary efficacy endpoint was change in A1C at 24 weeks and secondary efficacy endpoints included percentage of patients achieving A1C values <7% and ⩽6.5%, changes in FPG, body weight, β-cell function and fasting plasma insulin (FPI). At 24 weeks, the LS mean difference in A1C versus placebo was −0.56% (p < 0.001) and there was no difference in A1C change whether patients were receiving metformin or not. Significantly more patients in the lixisenatide group achieved an A1C value of <7% or ⩽6.5% compared with placebo (p < 0.001 for both). Lixisenatide also reduced FPG (LS mean difference −0.84 mmol/l, p < 0.0001) and slightly, though not significantly, reduced body weight (LS mean difference −0.41 kg, p = 0.1864) compared with placebo. β-Cell function at the end of 24 weeks was no different between lixisenatide- and placebo-treated patients (LS mean difference −0.25; 95% CI: −6.579, 6.070); however, decrease in FPI from baseline to 24 weeks was greater in the lixisenatide group (LS mean difference −9.36 pmol/L; 95% CI: −16.59 to −2.12). Postprandial glucose values and glucose excursions were not evaluated. The authors concluded that lixisenatide was an effective add-on to pioglitazone with or without concurrent metformin [Pinget et al. 2013].
Placebo comparators: use in patients receiving basal insulin
Lixisenatide has been studied in three trials that evaluated its use in patients with T2D receiving basal insulin. Patients in the GetGoal-Duo 1 study were already on a background of OADs and were initiated on insulin glargine that was systematically titrated over a 12 week period. Patients who still had an A1C ⩾ 7% at the end of the 12 week run-in phase were then randomized to receive either lixisenatide 20 μg once daily or placebo for 24 weeks while insulin titrations continued. The primary efficacy endpoint was change in A1C. Secondary efficacy endpoints included changes in weight and 2-hour PPG. Patients experienced a change in A1C of −1.0% with the addition of glargine and those who received lixisenatide experienced an additional 0.32% decrease compared with placebo (p < 0.0001). The LS mean difference in 2-hour PPG following a standardized meal test was −3.2 mmol/l compared with placebo (p < 0.0001). Body weight was also significantly reduced in the lixisenatide group compared with placebo (−0.9 kg, p = 0.0012). The study authors concluded that lixisenatide was an effective addon therapy to patients receiving OADs and insulin who needed additional glucose control. Lixisenatide decreased A1C and 2-hour PPG, and mitigated weight gain [Riddle et al. 2013b].
In the GetGoal-L study, patients with T2D with uncontrolled glucose values despite use of a stable dose of basal insulin (with or without a stable dose of metformin ⩾1500 mg per day) were randomized to receive lixisenatide 20 μg daily or placebo for 24 weeks. The primary efficacy endpoint was change in A1C from baseline and secondary efficacy endpoints included change in FPG, weight, insulin dose, self-measurement of plasma glucose (SMPG) and 2-hour PPG. At the end of 24 weeks, lixisenatide significantly decreased A1C (LS mean change −0.4%, p = 0.0002), body weight (LS mean change −1.3 kg, p < 0.0001), SMPG profile (LS mean difference −0.9 mmol/l, p < 0.0001) and 2-hour PPG (LS mean difference −3.8 mmol/l, p < 0.0001) compared with placebo. Daily basal insulin dose and FPG were not significantly different between lixisenatide and placebo at the end of 24 weeks. The authors concluded that lixisenatide offers an alternative to prandial insulin or other adjunctive agents and that it was an effective addon to basal insulin with or without concurrent metformin therapy [Riddle et al. 2013a].
The GetGoal-L-Asia study evaluated Asian patients with T2D with uncontrolled glucose values despite use of a stable dose of basal insulin with or without a stable dose of a sulfonylurea. These patients were randomized to receive lixisenatide in a stepwise dose increase to 20 μg daily or placebo for 24 weeks and the primary efficacy endpoint was change in A1C from baseline. Secondary efficacy endpoints included change in body weight, FPG, 2-hour PPG, SMPG profile and basal insulin dose. Patients treated with lixisenatide experienced a LS mean change in A1C of −0.88% compared with placebo (p < 0.0001). Lixisenatide also significantly improved 2-hour PPG and postmeal glucose excursions (LS mean difference −7.96 and −7.22 mmol/l, respectively; both p < 0.0001), SMPG profile (LS mean difference −1.35 mmol/l, p < 0.0001), FPG (LS mean difference −0.67 mmol/l, p = 0.0187) and basal insulin dose (LS mean difference −1.28 units, p = 0.0019). There was a trend toward weight loss with lixisenatide, but the difference compared with placebo was not statistically significant. The authors concluded that lixisenatide was an effective addon therapy to Asian patients receiving basal insulin with or without concurrent sulfonylurea therapy [Seino et al. 2012b].
Active comparators: comparison against a GLP-1 RA
Lixisenatide has been compared head-to-head with exenatide BID in a phase III study named GetGoal-X. In this study, lixisenatide produced similar reductions in A1C, better PPG reductions and less nausea compared with exenatide BID dosing.
The GetGoal-X study evaluated 634 patients in open-label fashion randomized to either lixisenatide 20 μg daily or exenatide 10 μg BID. Prior to randomization, patients were receiving a stable dose of ⩾1500 mg of metformin a day. The primary objective was a noninferiority assessment of change in A1C at 24 weeks. Other efficacy endpoints included change in FPG and weight. At 24 weeks, lixisenatide and exenatide demonstrated similar reductions in A1C with an LS mean difference of 0.17%, which met the noninferiority margin of 0.4%. Similar percentages of patients in each group achieved an A1C value <7% at 24 weeks. Decreases in FPG and body weight were similar for each group and not statistically different. Postprandial glucose data were not reported. The authors concluded that lixisenatide is a viable alternative to exenatide for improving glycemic parameters in a patient with T2D [Rosenstock et al. 2013].
Active comparators: comparison against rapid-acting prandial insulin
Lixisenatide has also been compared against the active comparator insulin glulisine in the GetGoal-Duo2 study. In this study, patients with T2D receiving background therapy of basal insulin with or without OADs were first assigned to a run-in phase in which all OADs except for metformin were discontinued and insulin glargine was optimized. At the end of the run-in phase, 894 patients with an A1C ⩾7% and ⩽9% were randomized to receive either lixisenatide 20 μg daily (n = 298) or insulin glulisine daily (GLU-1; n = 298) or three times daily (GLU-3; n = 298) for 26 weeks of therapy. The coprimary endpoints at the end of 26 weeks were change in A1C (noninferiority) and body weight (superiority). Secondary glycemic endpoints included change in FPG and 2-hour PPG at 26 weeks. Lixisenatide, GLU-1 and GLU-3 all reduced A1C from baseline to 26 weeks and lixisenatide was noninferior to both dosing schemes of insulin glulisine. The changes in A1C were −0.6% for lixisenatide and GLU-1, and −0.8% for GLU-3. The LS mean difference for lixisenatide versus GLU-1 was −0.05% (95% CI: −0.170 to 0.064%) and versus GLU-3 was 0.21% (95% CI: 0.095 to 0.328%). Lixisenatide demonstrated superior efficacy in weight reduction compared with GLU-3 at 26 weeks [LS mean difference −1.99 kg (95% CL: −2.593 to −1.396 kg)]. Change in FPG was similar and minimal across all three interventions, which is likely due to insulin glargine optimization during the run-in phase. Reductions in 2-hour PPG following a standardized breakfast were more marked with lixisenatide compared with either dosing scheme of insulin glulisine [−3.64 mmol/l (lixisenatide), −1.57 mmol/l (GLU-1), −1.41 mmol/l (GLU-3)]. The authors concluded that lixisenatide is a viable alternative to prandial insulin as an addon to basal insulin and metformin therapy [Rosenstock et al. 2015].
The GetGoal phase III clinical trial program demonstrated that lixisenatide effectively reduces A1C (range −0.7% to −1.0%), weight (range −0.2 to −2.96 kg) and 2-hour PPG (range −3.1 to −7.96 mmol/l) and does this across a variety of patient types. The main efficacy feature that distinguishes lixisenatide from other GLP-1 RAs is its ability to decrease 2-hour PPG for the meal immediately following injection, though lixisenatide has only been compared head-to-head with exenatide. Based on the marked PPG reductions produced by lixisenatide, a fixed ratio combination product of lixisenatide and insulin glargine (LixiLan – Lyxumia®/Lantus®) is currently being studied in phase III clinical trials.
Efficacy: special populations
A pooled analysis of 24-week efficacy data for elderly (⩾65 years old; n = 623) and very elderly (⩾75 years old; n = 79) patients enrolled in the GetGoal phase III clinical trial program demonstrated efficacy similar to younger patients. In patients <65 years old (n = 2565), the LS mean change in A1C compared with placebo was −0.56%. In patients ⩾65 and ⩾75 years old, the LS mean changes in A1C compared with placebo were −0.66% and −0.50%, respectively. The authors’ rationale for the persistence of lixisenatide efficacy in an elderly population is that its mechanism of glucose reduction relies on PPG reduction via gastric emptying more so than stimulation of insulin release from pancreatic beta cells [Raccah et al. 2015].
Lixisenatide has not been studied in phase III clinical trial specifically in patients with renal or hepatic impairment. The lixisenatide summary of product characteristics lists a general statement advising prescribers that there is ‘no dosage adjustment’ necessary in patients with hepatic impairment. A recent meta-analysis of patients with normal renal function (CrCl > 80 ml/min, n = 3515), mild renal impairment (CrCl 50–80 ml/min, n = 707) and moderate renal impairment (CrCl < 30 ml/min, n = 52) all experienced similar efficacy with lixisenatide with respect to reductions in A1C and 2-hour PPG values [Gomez-Huelgas et al. 2014].
Key safety considerations
Overall, lixisenatide appears to be well tolerated. Rates of common and/or clinically relevant AEs are given in Table 2. Like other GLP-1 RAs, the most common AEs are gastrointestinal (GI) in nature, mild and transient. Rates of nausea, the most common AE, ranged from 16.3% to 50% in patients taking lixisenatide in the phase III clinical studies [Fonseca et al. 2012; Seino et al. 2012b; Ahren et al. 2013; Yu Pan et al. 2014; Bolli et al. 2014; Pinget et al. 2013; Rosenstock et al. 2013, 2014; Riddle et al. 2013a, 2013b]. Despite the relatively large incidence of nausea, only 3.8–5% of patients receiving lixisenatide discontinued therapy for this reason [Seino et al. 2012b; Rosenstock et al. 2013, 2014; Bolli et al. 2014]. Rates of vomiting ranged from 6.7% to 18.2%. Using a one-step versus two-step titration or administering the drug in the morning or evening did not appear to have a significant or consistent impact on rates of GI AEs [Fonseca et al. 2012; Seino et al. 2012b; Ahren et al. 2013; Bolli et al. 2014]. In the head-to-head trial versus the GLP-1 RA exenatide, fewer patients reported nausea with lixisenatide compared with exenatide BID (24.5% versus 35.1%, p < 0.05) [Rosenstock et al. 2013]. In both groups, however, most cases of nausea resolved within the first 1 week.
Phase III clinical trials of lixisenatide: safety data.
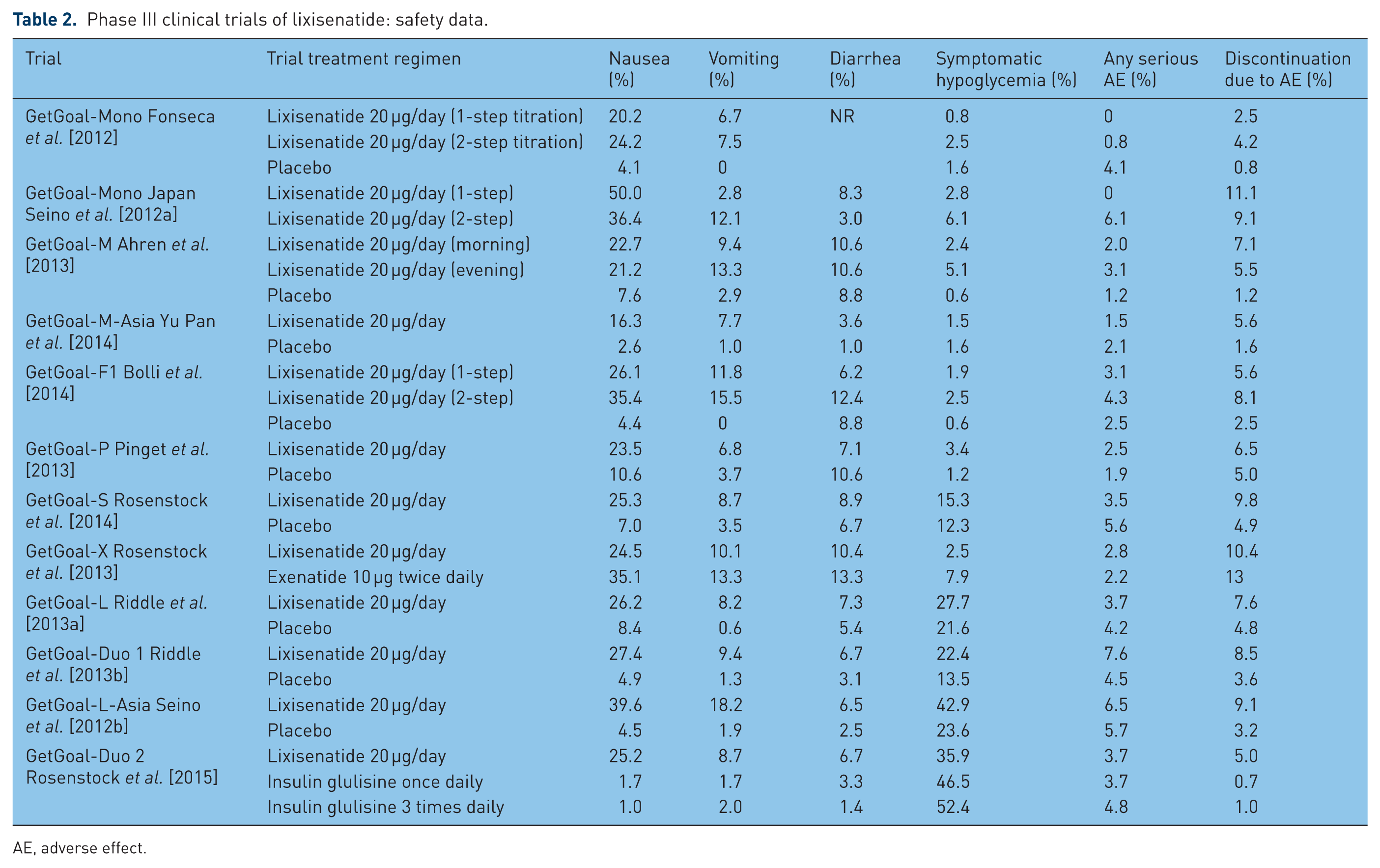
AE, adverse effect.
Like other GLP-1 RAs, lixisenatide also has a low risk of hypoglycemia. Rates of symptomatic hypoglycemia ranged from 0.8% to 3.4% in patients taking lixisenatide alone or in combination with metformin or pioglitazone [Fonseca et al. 2012; Seino et al. 2012b; Ahren et al. 2013; Bolli et al. 2014; Pinget et al. 2013; Rosenstock et al. 2013]. When added to basal insulin, rates of symptomatic hypoglycemia were higher (22.4–42.9%) and significantly more common than placebo [Riddle et al. 2013a, 2013b; Seino et al. 2012b]. In the head-to-head trial versus exenatide, patients taking lixisenatide had significantly fewer episodes of symptomatic hypoglycemia compared with patients taking exenatide BID (2.5% versus 7.9%, p < 0.05) [Rosenstock et al. 2014].
A concern with pancreatitis and use of GLP-1 RAs has been previously reported [Anderson and Trujillo, 2010]. The lixisenatide prescribing information states that a few cases of pancreatitis with its use have occurred but a causal relationship has not been established. Data from the Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) study demonstrated that there was no increase in the incidence of pancreatitis in patients receiving lixisenatide [Brigham and Women’s Hospital, 2015].
Safety considerations in special populations
Pooled analysis of patients enrolled in the phase III clinical trial program demonstrated that the incidence of GI AEs and symptomatic hypoglycemia were comparable between patients over the age of 65 and under 65 years [Raccah et al. 2015]. A meta-analysis of 9 of the phase III clinical trials revealed higher rates of GI AEs in patients with mild or moderate renal impairment compared with patients with normal renal function (27.8% versus 28.5% versus 19.4% respectively, p value not reported) [Gomez-Huelgas et al. 2014]. These trials included very few patients over 75 years of age (n = 79) and very few patients with moderate to severe renal dysfunction (n = 52), thereby limiting the generalizability to these patient populations.
The cardiovascular (CV) safety of lixisenatide was evaluated in patients with T2D and high cardiovascular risk in the ELIXA study [Brigham and Women’s Hospital, 2015]. This CV outcome study evaluated lixisenatide versus placebo in over 6000 patients with T2D who had recently experienced a spontaneous acute coronary syndrome event. Lixisenatide met the prespecified noninferiority criteria versus placebo for the composite primary endpoint of major adverse CV events (MACE) which included CV death, nonfatal myocardial infarction, nonfatal stroke and hospitalization for unstable angina [13.4% versus 13.2%; hazard ratio (HR) 1.02; 95% CI 0.89–1.17, p < 0.001). Additionally, no significant differences were observed for risk of heart failure or heart failure exacerbations.
Place in therapy
GLP-1 RAs are reasonable and appropriate addon therapies to metformin, sulfonylureas, pioglitazone and basal insulin in patients with T2D [American Diabetes Association, 2015; Handelsman et al. 2015]. The choice of which GLP-1 RA to select depends on patient and product factors. Lixisenatide is effective at lowering A1C and weight, and has shown superior effects when compared with placebo. It has demonstrated noninferiority to exenatide BID as well as insulin glulisine once daily and three times daily [Rosenstock et al. 2013, 2015]. It has not been studied against any other active comparator in the phase III clinical program. When directly compared with liraglutide in a phase IIb study, lixisenatide demonstrated superior effects on PPG but the clinical efficacy has not otherwise been directly compared with liraglutide or once weekly GLP-1 RAs [Meier et al. 2015]. It is important to know whether the improved effect on PPG translates to clinically meaningful reductions in A1C.
The shorter duration of action and distinct PD effect of lixisenatide on PPG support the use of this agent in combination with basal insulin. Four phase III studies were completed specifically evaluating this combination, including insulin-naïve patients and those on stable insulin doses. Lixisenatide was as effective as once or thrice daily insulin glulisine when added on to basal insulin without negative effects on weight and with lower rates of hypoglycemia [Rosenstock et al. 2015]. Therefore, lixisenatide could be considered as an alternative to rapid acting insulin analogs in patients on sufficiently titrated basal insulin who have achieved target fasting glucose levels but still have A1C levels between 7% and 8%. Lixisenatide should be more effective with basal insulin compared with longer acting GLP-1 RAs with less PPG effect, but only a head-to-head trial would answer that question.
Lixisenatide is well tolerated and has a more favorable AE profile compared with exenatide BID [Rosenstock et al. 2013]. It is administered once daily with a one-step increase to a single maintenance dose. In Europe, it is available in prefilled, 14-day, multi-use pen devices in either a 10 or 20 μg dose. User accuracy, ease-of-use and user preference of the lixisenatide pen device was compared with the exenatide BID and liraglutide pen devices in an open-label task and interview-based study [Stauder et al. 2014]. Tasks were completed faster (p < 0.001) with fewer errors (p = 0.001) with the lixisenatide pen compared with the exenatide pen. User satisfaction was higher with the lixisenatide and liraglutide pens compared with the exenatide pen (p < 0.001 for both). Although it is not available in the US to date, the acquisition cost in the European Union is lower than other GLP-1 RAs [Chaplan and Joseph, 2014].
Finally, current prescribing information in Europe indicates that no dose adjustment is needed for renal function or age. Lixisenatide can be used in patients with mild renal insufficiency (CrCl 50–80 ml/min), should be used with caution in patients with moderate renal insufficiency (CrCl 30–50 ml/min) and is not recommended in severe renal impairment (CrCl < 30 ml/min) due to a lack of data. Clinical experience in patients ⩾75 years of age is limited [Sanofi, 2014].
Submission to the US Food and Drug Administration (FDA) is planned for July 2015. In addition, phase III trials are ongoing evaluating a combination product containing lixisenatide and insulin glargine (LixiLan – Lyxumia®/Lantus®) [Zealand Pharma, 2015]. Regulatory submissions of LixiLan are expected to occur in the fourth quarter of 2015 (US) and in the first quarter of 2016 (Europe). Approval by the FDA, cost, and the design and ease of use of the delivery device will all be key factors influencing the ultimate place in therapy of this agent.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.