
Abstract
Background:
Myelofibrosis (MF) is a chronic myeloproliferative neoplasm characterized by progressive bone marrow fibrosis, splenomegaly, debilitating constitutional symptoms, and cytopenias. Despite the benefits of ruxolitinib in controlling symptoms and spleen volume, challenges such as myelosuppression and limited impact on underlying fibrosis persist, particularly in cytopenic patients. Flonoltinib maleate (FM), a novel JAK2/FLT3/CDK6 inhibitor, shows preliminary potential in improving hematologic parameters and reducing fibrosis.
Objectives:
To evaluate the efficacy of low-/high-dose FM compared with RUX (primary objective), along with safety and the pharmacokinetic profile of FM (secondary objectives), in patients with intermediate- to high-risk MF (Trial registration: NCT06457425).
Design:
FMF-02 is a multicenter, randomized, open-label, active-controlled, phase IIb clinical trial.
Methods:
Approximately 75 adults with primary or secondary MF will be randomized in a 1:1:1 ratio to receive low-dose FM (50 mg once daily), high-dose FM (100 mg once daily), or RUX (5, 15, or 20 mg twice daily, based on platelet count), with randomization stratified by the Dynamic International Prognostic Scoring System risk category. The primary endpoint is the proportion of patients achieving ⩾35% spleen volume reduction (SVR35) at week 24, as assessed by a blinded Independent Review Committee. Key secondary endpoints include the proportion with ⩾50% reduction in Total Symptom Score (TSS50), changes in myelofibrosis grade, objective remission rate [International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) criteria], and safety. Subjects in the RUX group who complete the 24-week treatment or experience disease progression due to splenomegaly will crossover to receive FM. All subjects will continue long-term therapy until meeting discontinuation criteria, followed by survival follow-up.
Results:
The study commenced in June 2024 and is currently ongoing. The results will provide comparative data on the efficacy and safety profiles of FM versus RUX, including analyses of spleen response, symptom burden, hematologic parameters, and bone marrow fibrosis.
Conclusion:
The FMF-02 trial is the first randomized phase IIb study directly comparing the novel inhibitor FM against RUX. Its findings are expected to generate pivotal evidence regarding whether FM offers superior or differentiated clinical benefits, thereby informing its potential as a frontline therapy for intermediate- to high-risk MF and guiding the design of future phase III studies.
Plain language summary
Myelofibrosis (MF) comprises primary MF (PMF) and secondary MF (including post-PV and post-ET MF). It features progressive bone marrow fibrosis, splenomegaly, debilitating constitutional symptoms, and cytopenias. The JAK inhibitors have revolutionized the management of MF, and ruxolitinib represents the mainstay of MF therapy, demonstrating superior improvements in symptom burden and spleen volume reduction. However, its use is often constrained by myelosuppressive toxicity and lack of disease-modifying effects, particularly in cytopenic patients. Flonoltinib maleate (FM) exhibits a different mechanism from ruxolitinib by simultaneously targeting JAK2, FLT3, and CDK6 with high selectivity, which play key roles in MF progression. Preclinical studies show FM inhibits the JAK-STAT pathway more potently than ruxolitinib, stabilizes platelets, and improves bone marrow fibrosis. In the phase IIa trial (NCT05153343), FM 100 mg resulted in a 35% or greater reduction in spleen volume (SVR35) in 81.8% of patients at week 24. The rates for best SVR35 and for a 50% or greater reduction in Total Symptom Score (TSS50) were 93.3% and 73.3%. Bone marrow fibrosis improvement occurred in 36.4% of patients. The FMF-02 is a multicenter, randomized, open-label, phase IIb clinical trial to compare the safety and effectiveness of FM and ruxolitinib phosphate (RUX) in patients with intermediate- to high-risk MF. Approximately 75 patients will be randomized 1:1:1 to low-dose FM (50 mg QD), high-dose FM (100 mg QD), or RUX (5/15/20 mg BID per platelet count). Investigators will track changes in spleen volume, symptom burden, myelofibrosis grade, objective remission rate, and safety over 48 weeks. Data from the FMF-02 study will provide key evidence on whether FM offers superior efficacy and safety compared to RUX, and describe the pharmacokinetics of FM, offering pivotal support for its potential as a frontline therapy in intermediate- to high-risk MF, and enabling phase III development.
Keywords
Introduction
Primary myelofibrosis (PMF), postpolycythemia vera myelofibrosis (PPV-MF), and postessential thrombocythemia myelofibrosis (PET-MF) are collectively referred to as BCR::ABL1-negative myeloproliferative neoplasm-associated myelofibrosis (MF). In European and American populations, MF incidence ranges from 0.47 to 1.98 per 100,000, predominantly affecting individuals over 40 years of age, with a slight male predominance. 1 Common clinical manifestations include peripheral blood abnormalities (leukocytosis, anemia, and thrombocytopenia), splenomegaly, extramedullary hematopoiesis, and associated complications. 2 Approximately 10%–20% of PMF patients progress to acute myeloid leukemia (AML) within 10 years of diagnosis. 3 The primary pathogenesis of MF involves constitutive activation of the JAK-STAT signaling pathway driven by mutations in JAK2 (predominantly V617F), MPL, and CALR. Patients classified as intermediate- or high-risk by scoring systems such as the Dynamic International Prognostic Scoring System (DIPSS) or DIPSS-Plus face substantially shortened survival and an elevated risk of leukemic transformation.4,5
The therapeutic landscape for intermediate- to high-risk MF has been revolutionized by JAK inhibitors, which form the cornerstone of treatment. Ruxolitinib (JAK1/JAK2 inhibitor) improves splenomegaly, symptoms, and overall survival but frequently causes dose-dependent anemia and thrombocytopenia, which may limit treatment intensity.6,7 Fedratinib (JAK2/FLT3 inhibitor) is approved for newly diagnosed or ruxolitinib-exposed patients. The JAKARTA trial showed that 47% of patients achieved ⩾35% spleen volume reduction at week 24, 8 while the gastrointestinal toxicity and Wernicke’s encephalopathy remain concerns.9,10 Pacritinib is a JAK2/IRAK1 inhibitor specifically approved for MF patients with severe thrombocytopenia (platelets < 50 × 10⁹/L). It reduces spleen volume and alleviates symptoms while uniquely improving anemia, addressing a critical limitation of conventional JAK inhibitors like ruxolitinib that exacerbate cytopenias in this high-risk subgroup. 11 Nevertheless, its overall efficacy in nonthrombocytopenic patients remains inferior to ruxolitinib. 2 Momelotinib, a JAK1/2 and ACVR1 inhibitor, addresses anemia in MF patients,12,13 significantly improved anemia-related endpoints and symptoms compared with danazol in symptomatic, 12 but its splenic response rates remain modest compared to ruxolitinib, and it carries risks of neuropathy and hypertension. 2 Although effective in reducing splenomegaly and alleviating constitutional symptoms, most approved agents exhibit significant limitations in cytopenic patients and lack disease-modifying activity. 14–16 Many MF patients present with or develop cytopenias that preclude optimal dosing of existing agents. Hence, there is a pressing need for next-generation therapies that can address both symptom burden and disease biology while maintaining tolerability in cytopenic or JAK inhibitor–intolerant populations.
JAK2V617F disrupts the autoinhibitory pseudokinase domain (JH2), leading to constitutive activation of JAK2 kinase activity and STAT-mediated activation of transcription. 17 MPLW515 results in constitutive activation of TPO-receptor signaling, JAK2 phosphorylation, and activation of STAT-dependent transcription. 18 CALR mutation drives constitutive JAK2 phosphorylation, and STAT transcriptional activation via MPL dependence, and JAK2 inhibition is effective in patients with CALR-mutated MF. 19 Ruxolitinib and fedratinib bind to the kinase domain (JAK2 JH1), which likely contributes to their insufficient selectivity and resultant severe side effects.20–22 Bristol-Myers Squibb (BMS) has developed a highly selective TYK2 inhibitor named BMS-986165. Unlike other JAK inhibitors, this drug binds to the pseudokinase domain (JH2), inducing a conformational change. This mechanism has been shown to significantly enhance selectivity and reduce adverse effects. 23 Furthermore, CDK6 expression is markedly upregulated in hematopoietic progenitor cells from MF patients. 24,25 CDK6 inhibitors offer a promising therapeutic strategy for MF by targeting dysregulated cell cycle progression in malignant hematopoietic stem/progenitor cells.24,26 Preclinical studies (e.g., palbociclib) demonstrate reductions in splenomegaly and fibrosis, effects not observed with ruxolitinib. 24 Clinical trials are currently underway investigating the combination of CDK4/6 inhibitors with RUX for the treatment of myelofibrosis (ClinicalTrials.gov: NCT05714072).
Flonoltinib maleate (FM), a novel oral JAK2/FLT3/CDK6 inhibitor, uniquely targets these pathogenic axes. Biochemical kinase assays demonstrated potent inhibition of JAK2 (IC50 = 0.8 nM), with >600-fold selectivity over JAK1/JAK3, alongside meaningful activity against FLT3 (IC50 = 15 nM) and CDK6 (IC50 = 35 nM). The high selectivity of JAK2 is achieved by binding to both JH2 and JH1 simultaneously, with a stronger binding ability to JH2 than to JH1. 27 Preclinically, FM has a better ability to inhibit the JAK-STAT signaling pathway than ruxolitinib and fedratinib, and effectively suppresses JAK2V617F-driven splenomegaly and reduces fibrosis. 27 These findings support its potential as a disease-modifying agent, in contrast to first-generation JAK inhibitors that primarily offer symptom control. A completed multicenter phase I/IIa study (NCT05153343) evaluating FM at doses ranging from 25 to 225 mg in 30 patients with MF or other advanced MPNs has demonstrated encouraging clinical efficacy. Among the 15 evaluable MF patients treated at the 100 mg dose level, FM achieved a ⩾35% spleen volume reduction (SVR35), the best SVR35, and ⩾50% reduction in Total Symptom Score (TSS50) of 81.8%, 93.3%, and 73.3%, respectively, at week-24 in MF patients. Bone marrow fibrosis improvement was also observed in 36.4% MF patients. Notably, early signals of anemia and thrombocytopenia reversal were observed in cytopenic patients, including transfusion-dependent individuals. These preliminary findings suggest potential benefit in patients with advanced disease or suboptimal tolerance to existing JAK inhibitors. FM also demonstrated a manageable safety profile, with the most common treatment-emergent adverse events (AEs) being hematologic (e.g., thrombocytopenia, anemia, neutropenia), mostly grades 1–2 in severity. No dose-limiting toxicities (DLTs) were reported. Pharmacokinetic profiling indicates rapid oral absorption (Tmax, 2–4 h), long half-life (~16–38 h), and high systemic exposure, comparable to 240 mg fedratinib at doses as low as 50 mg. The steady-state trough concentration (Cmin, ss) of free FM in plasma at 50 and 100 mg doses significantly exceeded: (1) the threshold required for inhibiting JAK2, FLT3, and CDK6 kinase activities; and (2) the concentration suppressing proliferation in JAK2V617F mutant cell lines. Given the favorable therapeutic window, differentiated mechanism, and emerging clinical data, FM is well-positioned for further clinical evaluation in MF, particularly in cytopenic or JAK inhibitor–intolerant populations. Among JAK inhibitors, RUX is the most widely used medication for the treatment of MF, demonstrating excellent efficacy in alleviating symptom burden and reducing spleen volume. Therefore, RUX has been selected as the comparator drug in the clinical trial for FM.
This trial is the first randomized, controlled phase IIb study directly comparing FM versus ruxolitinib phosphate (RUX) in intermediate- to high-risk MF patients, including those with low baseline platelet counts. Approximately 75 patients will be enrolled and randomized in a 1:1:1 ratio to receive either oral low-dose FM, oral high-dose FM, or oral RUX. This trial aims to assess whether FM can offer improved clinical benefit, including the efficacy and safety over RUX in MF patients with limited treatment options, particularly those with cytopenias at baseline. Additionally, we will perform subgroup analyses to assess differences in efficacy between patients with JAK2V617F mutation versus wild-type status. The results of this trial may inform the future treatment paradigm and support the use of FM in broader patient populations, particularly those underserved by existing therapies.
Methods and analysis
Design
This multicenter, open-label, randomized, active-controlled phase IIb clinical trial (ClinicalTrials.gov: NCT06457425, https://clinicaltrials.gov/study/NCT06457425) conducted in China aims to evaluate the efficacy and safety of FM versus RUX in patients with intermediate-2 or high-risk MF, as defined by the DIPSS. The study flow diagram is presented in Figure 1.

Phase IIb trial flowchart.
The study was initiated at 33 research centers in China. Key participating sites include the Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences, and West China Hospital of Sichuan University, etc. Approximately 75 patients will be enrolled and randomized in a 1:1:1 ratio to receive oral either low-dose FM (50 mg once daily), high-dose FM (100 mg once daily), or RUX (5, 15, or 20 mg twice daily, based on platelet counts per prescribing information). The calculation assumed SVR35 rates of ~70% for FM and ~30% for RUX in Chinese myelofibrosis patients. Using PASS software (two-sided test), approximately 20 patients per arm provided an estimated 80% power to detect this difference. To account for a potential 20% dropout rate, the target was increased to 25 per arm (75 total). This study comprises a core treatment phase of 24 weeks, followed by an extended treatment phase. Randomization will be stratified by DIPSS risk category (intermediate-2 vs high). For the core treatment phase (weeks 0–24), all randomized participants will receive study treatment for 24 weeks. Participants who experience disease progression due to nonsplenomegaly-related causes (defined as bone marrow blasts > 20%; or peripheral blood blasts ⩾20% with an absolute blast count ⩾1 × 10⁹/L, sustained for at least 2 weeks) 28 during this period will discontinue treatment and withdraw from the study. Participants with disease progression manifested as splenomegaly (defined as ⩾25% spleen volume increase from baseline by CT/MRI, yet not meeting the 2013 UMNET/IWG-MRT consensus criteria) could continue treatment for 4–8 additional weeks or until the next scheduled disease assessment if the investigator deemed continued therapy clinically appropriate. A repeat spleen assessment using CT or MRI will be conducted and reviewed by an Independent Review Committee (IRC). If confirmed splenic progression is observed, FM-treated patients (both low- and high-dose arms) will discontinue study drug, while patients in the RUX arm will proceed to the extended phase. This study was designed as a single-stage phase IIb investigation. Participants receiving FM who complete the 24-week core treatment phase without progression will continue their assigned dose in the extended phase treatment phase (week 25 onwards), followed by an additional 24-week crossover phase to further explore the efficacy and tolerability of FM between weeks 24 and 48. Patients in the RUX arm who complete 24 weeks without progression, as well as those who experience splenomegaly-driven progression during the core phase, will undergo a second randomization (1:1) to receive either FM 50 or 100 mg once daily. Concomitant use of RUX is prohibited in patients assigned to FM treatment. All subjects will continue long-term therapy until meeting discontinuation criteria, and all participants who complete treatment or withdraw from the study will undergo a 4-week safety follow-up following the last dose of investigational drugs.
Objectives and endpoints
The FMF-02 study aims to comprehensively evaluate the efficacy, safety, and pharmacokinetics of FM in patients with intermediate-2 or high-risk MF, compared to RUX as an active control (Box 1). The key objective is to demonstrate the potential of FM as a novel therapeutic option for patients with cytopenic phenotypes, a subgroup with limited treatment alternatives under current JAK inhibitor-based regimens. In addition to assessing spleen volume reduction and symptom improvement, this study also evaluates hematologic responses, bone marrow fibrosis, and pharmacokinetic characteristics of FM.
Study objectives.

The primary efficacy endpoint is the proportion of participants achieving a ⩾35% reduction in spleen volume from baseline at week 24, as assessed by a blinded independent radiology committee (IRC; Box 2). Secondary efficacy endpoints include spleen volume reduction at additional timepoints (weeks 12, 36, and 48), assessed by both investigators and the IRC; time to first spleen response (TTR); best spleen response rate; duration of maintained spleen response (DoMSR); and time to spleen volume re-enlargement. Symptom-related endpoints include the proportion of participants achieving ⩾50% reduction in MPN-SAF TSS total symptom score, as well as change from baseline in TSS over time. Additional endpoints include the proportion of participants with stable or improved bone marrow fibrosis grading and the objective response rate (ORR) based on the IWG-MRT consensus criteria. Safety endpoints include the incidence and severity of AEs and serious AEs (SAEs), changes in laboratory parameters (hematology, biochemistry, coagulation, urinalysis, and pregnancy testing), vital signs, Eastern Cooperative Oncology Group (ECOG) performance status, 12-lead electrocardiogram (ECG), physical examination findings, and chest X-ray results.
Study endpoints.

Patient eligibility and recruitment
Eligible participants are adult patients (aged 18–75 years) with a confirmed diagnosis of PMF, PPV-MF, or PET-MF, classified as intermediate-2 or high-risk according to the DIPSS. Patients must present with symptomatic splenomegaly, have an ECOG performance status of 0–2, and meet prespecified hematologic and organ function criteria. All participants are required to provide written informed consent before enrollment. Patients with documented resistance or intolerance to prior ruxolitinib therapy, or prior use of any JAK inhibitor within 4 weeks before first dosing, or significant comorbidities that could interfere with study participation will be excluded. The full inclusion and exclusion criteria are summarized in Table 1.
Patient eligibility criteria.

DIPSS, dynamic international prognostic scoring system; ECOG, Eastern Cooperative Oncology Group; EOT, End of treatment; MF, Myelofibrosis; PET-MF, postessential thrombocythemia myelofibrosis; PMF, Primary myelofibrosis; PPV-MF, postpolycythemia vera myelofibrosis.
Recommended data collection and primary analyses
The overall study schedule, including screening, treatment, and follow-up visits, is summarized in the visit schedule table (Table 2). Baseline demographic and clinical characteristics will be collected at the time of enrollment, including disease history, risk stratification, prior treatments, and laboratory values. Throughout the trial, participants will undergo routine assessments per protocol, including physical examination, ECOG performance status, laboratory tests (hematology, chemistry, coagulation, urinalysis), 12-lead ECG, and symptom evaluation using the MPN-SAF TSS scale. All imaging-based spleen volume assessments will be conducted using MRI or CT, and independently evaluated by an IRC. Gene mutation testing included JAK2V617F (quantitative and qualitative), CALR (qualitative), MPLW515L/K (qualitative), and BCR::ABL fusion gene testing. At the EOT visit, quantitative JAK2V617F testing was performed only in patients who were JAK2V617F positive at baseline. Bone marrow aspiration and biopsy include bone marrow cytological analysis (bone marrow smear), reticulin (silver impregnation) stain, chromosome karyotype analysis (peripheral blood samples may be used if a “dry tap” occurs during bone marrow aspiration), and pathological biopsy. Bone marrow smear examination is performed when blasts account for ⩾20% on the peripheral blood smear. Bone marrow aspiration/biopsy is only required at screening, 12, 24, and 48 weeks.
Study schedule.
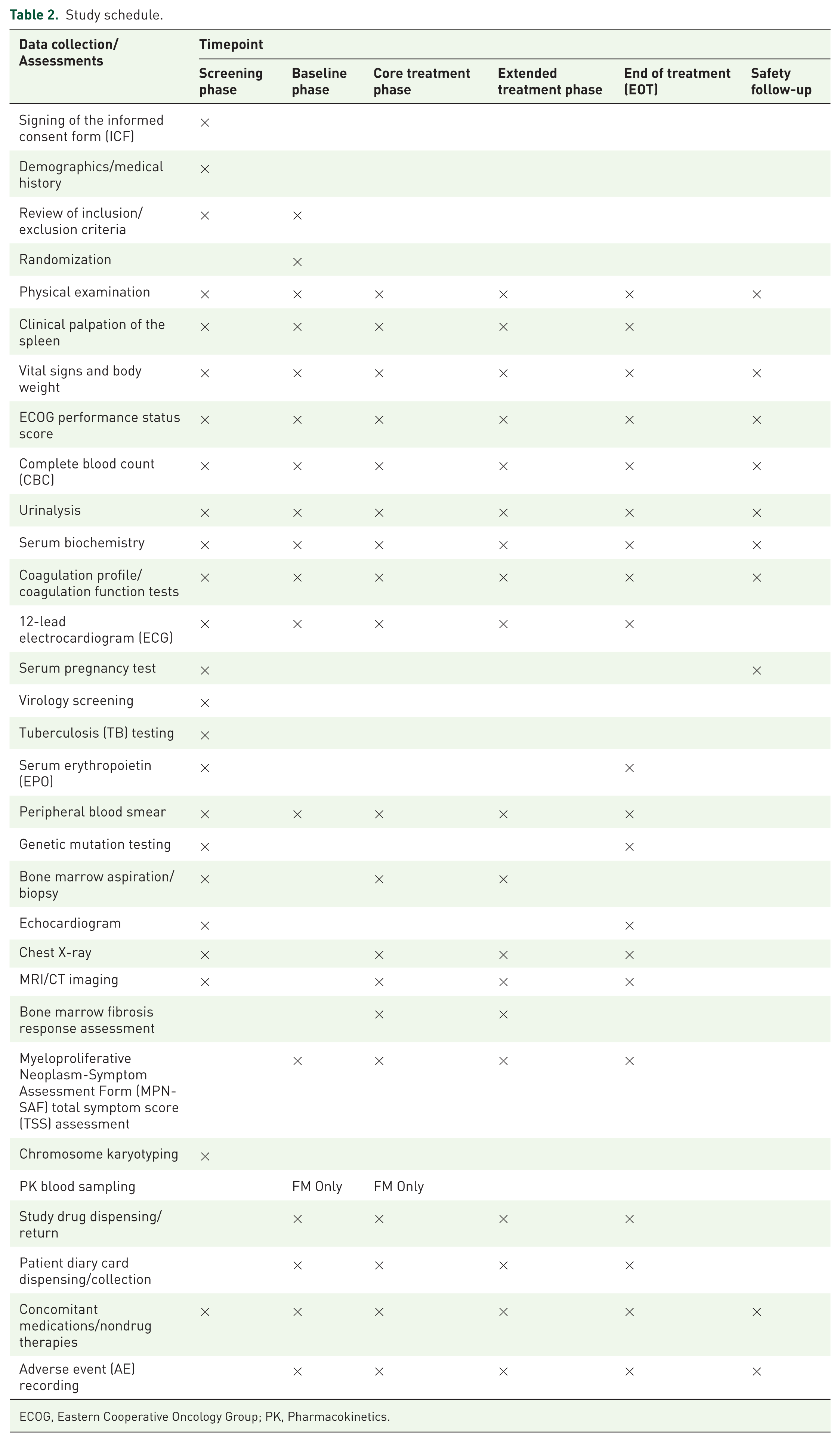
ECOG, Eastern Cooperative Oncology Group; PK, Pharmacokinetics.
Primary efficacy analyses will focus on the proportion of participants achieving SVR35 from baseline at week 24, as assessed by a blinded Independent Review Committee, and measures spleen volume using CT/MRI. For participants who discontinue early or experience disease progression or death before week 24, a composite strategy will be employed to classify such cases as nonresponders. For other nonprogression-related discontinuations, a hypothetical strategy using multiple imputation under a missing-at-random (MAR) assumption will be applied. Secondary analyses will include evaluation of spleen response at additional timepoints (weeks 12, 36, and 48), spleen response duration (DoMSR), time to spleen response, best spleen response, and changes in TSS scores over time. Time-to-event data such as DoMSR and duration of sustained response will be analyzed using the Kaplan–Meier method. Symptom burden will be assessed using changes from baseline in MPN-SAF TSS scores, analyzed using mixed-effects models for repeated measures (MMRM). Bone marrow fibrosis grading will be assessed at baseline and follow-up bone marrow biopsies (weeks 12, 24, and 48), based on central pathology review.
Pharmacokinetic (PK) sampling will be conducted in a subset of patients receiving FM, with peripheral blood samples collected at specified timepoints postdose. For the first six subjects in FM 50 and 100 mg groups, peripheral blood samples will be collected at the following timepoints, including Day 1 (D1): Within 1 h pre-dose; 0.5 h ± 2 min, 1 h ± 2min, 2 h ± 3 min, 3 h ± 3 min, 4 h ± 5 min, 6 h ± 7 min, 8 h ± 10 min, 12 h ± 15 min, and 24 h ± 30 min postdose; Week 2: Within 1 h predose; 0.5 h ± 2 min, 1 h ± 2 min, 2 h ± 3 min, 3 h ± 3 min, 4 h ± 5 min, 6 h ± 7 min, 8 h ± 10 min, 12 h ± 15 min, and 24 h ± 30 min postdose; Weeks 4, 12, and 24: Within 1 h predose. Noncompartmental methods will be used to derive key pharmacokinetic parameters, including Cmax, Tmax, MRT, AUC0–24h, AUC0-∞, and t1/2. Descriptive statistics (mean, SD, CV, median, min, max) will be reported for each PK parameter, and all below quantification limit (BQL) values will be handled per prespecified rules.
All data will be collected using an electronic data capture (EDC) system, with real-time query resolution and data validation performed by site investigators and the sponsor. Clinical monitoring, centralized imaging review, and data management will be conducted in accordance with Good Clinical Practice (GCP).
Safety
The safety of all treatments will be assessed based on the incidence, type, severity, and causality of AEs and SAEs, which will be coded using MedDRA terminology and graded according toNational Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 5.0. Any serious or unexpected safety events will be reported promptly to the Institutional Review Board/Independent Ethics Committee (IRB/IEC) in accordance with applicable regulatory and ethical requirements. Safety analyses will be performed on all participants who receive at least one dose of study medication, and all safety data will be analyzed according to the actual treatment received.
Statistical methods
All statistical analyses will be performed using SAS® for Windows (version 9.4 or higher) and Phoenix WinNonlin® (version 8.1 or higher) for pharmacokinetic calculations, in accordance with ICH E9 guidelines. Descriptive statistics for continuous variables will include the mean, standard deviation (SD), median, interquartile range, minimum, and maximum. For categorical variables, absolute and relative frequencies will be presented in contingency tables. Time-to-event data will be summarized using Kaplan–Meier methods, with 95% confidence intervals for medians calculated using the Brookmeyer–Crowley method. Further statistical details will be specified in a separate statistical analysis plan (SAP) finalized prior to database lock.
The full analysis set (FAS) will include all randomized participants who received at least one dose of study drug, analyzed according to the intention-to-treat principle, and will serve as the primary analysis population for efficacy endpoints. The safety set (SS) will comprise all participants who received at least one dose of the study drug and have any post-baseline safety data, and will be used for safety analyses. Pharmacokinetic analyses will be conducted on the PK concentration set (PKCS) and PK parameter set (PKPS), based on predefined inclusion criteria for evaluability.
The primary efficacy endpoint is the proportion of participants achieving ⩾35% spleen volume reduction from baseline at week 24 (assessed by IRC). Proportions and their two-sided 95% confidence intervals will be estimated using the Clopper–Pearson method. Between-group comparisons (including subgroup analyses) will be evaluated using exact tests, with Cochran–Mantel–Haenszel (CMH) chi-square tests adjusted for baseline stratification factors as sensitivity analyses. Handling of intercurrent events will follow a composite strategy or hypothetical strategy, depending on the event type. For participants who discontinue treatment due to nondisease progression events, missing data will be handled using multiple imputation under the missing-at-random (MAR) assumption, with baseline and postbaseline spleen volume data and treatment arm as covariates. Secondary efficacy endpoints include spleen volume response at other timepoints, symptom improvement, fibrosis grading, and objective remission rate. Binary endpoints will be summarized similarly to the primary analysis, without imputation for missing values (nonresponse imputation applied). Time-to-event variables such as spleen response duration will be analyzed using Kaplan–Meier estimates. Longitudinal symptom scores will be analyzed using MMRM, estimating least-squares mean changes from baseline and between-group differences with 95% confidence intervals.
Safety will be evaluated descriptively within the SS population, including adverse events, laboratory tests, ECGs, vital signs, and physical examinations. PK analyses will use noncompartmental methods with parameters summarized using both arithmetic and geometric means. No adjustment for multiplicity will be applied to secondary or exploratory endpoints. No formal interim analysis is planned.
Patient and public involvement
There was no patient or public involvement in the design or implementation of the FMF-02 study. Study results will be shared with participants through the investigators following database lock and final analysis.
Discussion
Patients with primary or secondary MF face reduced life expectancy. Among JAKi, RUX represents the mainstay of MF therapy, demonstrating superior improvements in symptom burden and spleen volume reduction. 29 However, many patients cannot tolerate RUX mainly due to drug-related cytopenias, and even patients with an initial response often develop resistance after 2–3 years of therapy. 30 Consequently, novel mechanism-based therapies are urgently needed for MF patients. FM offers a differentiated mechanism of action by targeting JAK2, FLT3, and CDK6 with high selectivity, potentially addressing anemia and thrombocytopenia, which are common in advanced MF. Unlike currently marketed JAK2 inhibitors such as RUX that bind exclusively to the JH1 (kinase domain), FM simultaneously engages both the JH1 and autoinhibitory JH2 pseudokinase domains of JAK2. 27 This unique dual-domain mechanism enhances selectivity for mutant JAK2V617F while minimizing off-target effects on JAK1/JAK3, potentially mitigating cytopenias associated with broad JAK-STAT pathway inhibition. Targeting CDK6, a master regulator of hematopoietic stem/progenitor cell proliferation and fibrogenic signaling, provides a direct strategy to attenuate bone marrow fibrosis. 31 Preclinical evidence demonstrates that CDK6 targeting via either palbociclib or genetic ablation reduces NF-KB and TGF-β signaling pathway-driven stromal activation and collagen deposition, conferring FM with enhanced antifibrotic activity, a critical unmet need unaddressed by current JAK inhibitors.24,26,31 Furthermore, FLT3 signaling promotes extramedullary hematopoiesis and leukemic transformation in myelofibrosis. 32 It may provide additional protection against disease progression, particularly in high-risk molecular subgroups. Thus, the FMF-02 trial was designed to evaluate the efficacy and safety of FM, a novel, JAK2/FLT3/CDK6 selective inhibitor, in comparison with RUX.
In preclinical, FM demonstrates superior JAK-STAT pathway inhibition versus RUX, 27 and can stabilize platelet levels and improve bone marrow fibrosis via suppressing the TGF-β signal pathway. In the phase IIa trial (NCT05153343), the oral, once-daily dosing and manageable toxicity profile observed in MF patients make it an attractive alternative for patients who are either intolerant to or suboptimally managed with RUX. FM 100 mg achieved SVR35, best SVR35, and ⩾50% reduction in TSS50 of 81.8%, 93.3%, and 73.3%, respectively, at week-24 in MF patients and manageable toxicities. Bone marrow fibrosis improvement was also observed in 36.4% MF patients. Subgroup analysis showed no statistically significant difference in spleen response between prior JAKi-exposed and JAKi-naïve patients: Week-24 SVR35 rates were 70.0% (7/10) versus 83.3% (10/12), respectively. 33 Given the increasing emphasis on tailored therapies and the emergence of molecularly defined subgroups in MF, FMF-02 also integrates optional biomarker and pharmacokinetic assessments to explore response predictors. FM comparison with RUX, rather than a placebo or hydroxyurea control, reflects the evolving treatment paradigm and underscores the trial’s aim to position FM within the context of current standard-of-care therapies, enabling a clinically meaningful assessment of its potential superiority in selected patient populations.
As a highly selective next-generation JAK2 inhibitor with favorable tolerability in cytopenic patients, FM has the potential to redefine the treatment paradigm for myelofibrosis. By directly challenging RUX, the current standard-of-care and cornerstone therapy, this study aims to position FM as a new frontline backbone for intermediate- to high-risk patients, especially those with cytopenic phenotypes inadequately served by existing therapies. In parallel, it may offer a much-needed option in second-line settings, including patients who are refractory, resistant, or intolerant to RUX, the current standard among JAK2 inhibitors. The biological and therapeutic heterogeneity among MF patients with different driver mutations (JAK2V617F, CALR, and MPL) is a crucial consideration. We will conduct a subgroup analysis of efficacy between those with JAK2V617F, CALR, and MPLW515L/K mutations and those without mutations. Preclinically, FM induced apoptosis and inhibited colony formation in primary cells from CALR-mutated MF patients in a concentration-dependent manner. As of July 2025, in the phase IIb clinical trial, patients with CALR mutations at week 24 achieved significantly higher SVR35 and TSS50 response rates in the FM treatment groups compared to the RUX group. Specifically, SVR35 was reached in 7 of 8 (87.5%) CALR-mutated patients treated with FM, versus 0 of 2 in the RUX arm. 34 Similarly, TSS50 was achieved in 8 of 8 (100%) such patients on FM groups, compared with 1 of 2 on RUX (week 12). However, we did not explore the effects of FM on MPL-mutant MF mouse models or primary cells from MF patients in preclinical studies, and no MPL-mutant MF patients have been found among the patients currently enrolled. In the future, we will also focus on the efficacy of FM in treating MPL-mutated MF mouse models and clinical patients. Additionally, for patients who underwent bone marrow reticulin (silver impregnation) staining, fibrosis grading comparisons will be performed to determine the effects of FM and RUX on bone marrow fibrosis. Data from this trial will provide critical evidence to support FM’s potential to reshape clinical decision-making and address key unmet needs across the MF treatment continuum.
FMF-02 is an ongoing, multicenter, randomized, phase IIb clinical trial conducted in China. The trial opened to recruitment in Q2 2024 using protocol version 1.0 (dated December 1, 2023). The current version in use is Version 2.1 (April 18, 2025, detailed in the “Supplemental Material (protocol Ver2.1)”). As of July 2025, all 75 patients have been successfully enrolled across 33 sites spanning multiple regions of China. A total of 43 participants completed the week 12 assessment, and 25 completed the week 24 assessment. Participants continue to receive either FM or RUX per randomization, with follow-up ongoing according to the prespecified schedule. While the FMF-02 trial will provide important comparative data on a novel therapeutic agent, several limitations should be acknowledged. First, the study was conducted exclusively in Chinese patients, which may limit the generalizability of the findings to other ethnic or geographic populations until confirmed in broader, global studies. Second, the open-label design introduces the potential for assessment bias, particularly for subjective endpoints such as symptom scores; however, the use of a blinded IRC for the primary imaging-based endpoint (spleen volume reduction) mitigates this concern for the key efficacy measure. Third, as an exploratory phase IIb study, the sample size was not formally powered for hypothesis testing (superiority or noninferiority) but was chosen based on practical considerations to generate preliminary efficacy and safety signals. Consequently, the statistical power to detect differences, especially in subgroup analyses (e.g., by mutation status), is limited. Finally, the 24-week primary assessment period for spleen response is standard in MF trials, but longer follow-up is needed to fully characterize the durability of response, the impact on bone marrow fibrosis, and long-term safety. Despite these limitations, the FMF-02 study will offer valuable head-to-head evidence that will inform the design of a subsequent phase III confirmatory trial.
Conclusion
Although the JAK inhibitors have revolutionized the management of intermediate- to high-risk myelofibrosis, first-generation agents such as ruxolitinib remain constrained by myelosuppressive toxicity and limited disease-modifying activity, particularly in cytopenic patients. The critical unmet need for agents that simultaneously target symptom burden, disease biology, and cytopenic phenotypes underpins the FMF-02 trial. As the first randomized phase IIb study directly comparing the novel JAK2/FLT3/CDK6 inhibitor FM against RUX, this investigation aims to determine whether FM provides superior clinical benefit through efficacy-enhanced, improved hematologic tolerability, and disease-modifying potential. Results will provide pivotal evidence supporting FM’s potential positioning as frontline therapy for intermediate- to high-risk MF while strategically laying groundwork for phase III development.
Supplemental Material
sj-docx-1-tah-10.1177_20406207261424845 – Supplemental material for Assessment of flonoltinib maleate versus ruxolitinib phosphate in intermediate- to high-risk myelofibrosis (FMF-02): study protocol for a multicenter, randomized, open-label phase IIB trial
Supplemental material, sj-docx-1-tah-10.1177_20406207261424845 for Assessment of flonoltinib maleate versus ruxolitinib phosphate in intermediate- to high-risk myelofibrosis (FMF-02): study protocol for a multicenter, randomized, open-label phase IIB trial by Linyu Yang, Ke Tan, Rui Liang, Wei Zhang, Yiqun Wang and Lijuan Chen in Therapeutic Advances in Hematology
Supplemental Material
sj-docx-2-tah-10.1177_20406207261424845 – Supplemental material for Assessment of flonoltinib maleate versus ruxolitinib phosphate in intermediate- to high-risk myelofibrosis (FMF-02): study protocol for a multicenter, randomized, open-label phase IIB trial
Supplemental material, sj-docx-2-tah-10.1177_20406207261424845 for Assessment of flonoltinib maleate versus ruxolitinib phosphate in intermediate- to high-risk myelofibrosis (FMF-02): study protocol for a multicenter, randomized, open-label phase IIB trial by Linyu Yang, Ke Tan, Rui Liang, Wei Zhang, Yiqun Wang and Lijuan Chen in Therapeutic Advances in Hematology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.