
Abstract
Background:
Detection of measurable residual disease (MRD) is becoming the standard of care in the prognostication of acute myeloid leukaemia post-chemotherapy, but its role in early post-haematopoietic stem cell transplantation (HSCT) is less defined.
Objectives:
This study aims to examine the role of MRD detection by molecular tools in early post-HSCT settings and its significance in prognostication among other clinicopathologic factors.
Design:
It is a retrospective cohort study.
Methods:
We examined MRD early post-HSCT (median: 26 days) in patients receiving allogeneic HSCT at complete remission by droplet digital PCR or next-generation sequencing targeting leukaemia-associated mutations. The effect of MRD positivity and other clinicopathologic variables on post-HSCT leukaemia-free survival (LFS), overall survival (OS) and cumulative incidence of relapse (CIR) were investigated.
Results:
One hundred fifty-nine patients were included, and the median follow-up time was 6 years. Early post-HSCT MRD positivity was observed in 36% patients and was associated with higher CIR (37.1% vs 13.2% at third year, p = 0.0005), inferior LFS (median: 3.8 years vs not reached, p = 0.002) and OS (median: 10.6 years vs not reached, p = 0.019). It was the only significant factor associated with inferior post-HSCT LFS, CIR and OS in both univariate and multivariate analyses.
Conclusion:
This study demonstrated that early MRD positivity was predictive of a higher risk of relapse, and inferior LFS and OS post-HSCT. This information laid the foundation for designing MRD-guided strategies early post-HSCT to prevent haematological relapse.
Introduction
Acute myeloid leukaemia (AML) is a group of heterogeneous diseases with distinct clinicopathologic, cytogenetic and genetic characteristics, sharing in common an abnormal increase in blasts in peripheral blood (PB) or bone marrow (BM).1–3 For young and fit patients, induction and consolidation chemotherapy are the mainstays of treatment. They are risk stratified according to the specific cytogenetics and genetic mutations at diagnosis.4–7 The prognostication informs decisions on allogeneic haematopoietic stem cell transplantation (allo-HSCT) at first complete remission (CR1). However, disease relapse post-HSCT remains a major cause of treatment failure. 8 Once haematological relapse has occurred, treatment options are very limited8–11 and prognosis is generally dismal.4,8,12,13 There is an unmet clinical need to predict relapse early post-HSCT so that preventive measures can be implemented.
Measurable residual disease (MRD) detection in AML has become a useful tool for prognostication and disease monitoring. Early detection of MRD post-HSCT is of therapeutic relevance as it may allow manipulation of allo-immunity and enhancement of graft-versus-leukaemia (GVL) activity to prevent frank haematological relapse. Multi-parametric flow cytometry (MFC) has been used, but it is labour-intensive and requires a high level of expertise in flow cytometry.14,15 Recent advances in next-generation sequencing (NGS) and droplet digital PCR (ddPCR) have enabled more robust and automated detection of MRD.16–18 Moreover, the design of a patient-specific MRD platform is possible, particularly in patients whose gene mutations are private. In this study, we examined the role of MRD detection by molecular tools in early post-HSCT settings and its significance in prognostication among other clinicopathologic factors.
Methods
Patients and treatment
Adult AML patients who received allo-HSCT at CR at Queen Mary Hospital, Hong Kong, from 2003 to 2023 and whose AML showed mutations amenable to ddPCR detection (Table S1) or targeted sequencing based on NGS were included. One patient included in the study received allo-HSCT in another hospital outside Hong Kong and continued post-HSCT care at our centre. Patients who received HSCT with persistent disease or at relapse were excluded. Patients who were ⩽55 years old and fit received myeloablative conditioning (MAC), and those who were unfit or >55 years old received reduced intensity conditioning. Table S2 summarised the conditioning regimens under different conditions. All patients received standard anti-microbial and graft-versus-host disease prophylaxes as described previously. 13 Specifically, cyclosporine A and methotrexate were used in HSCT from matched sibling donors. Cyclosporine A, methotrexate and mycophenolate mofetil were used in those from matched unrelated donors and mismatched unrelated donors before mid-2019. Post-transplant cyclophosphamide, cyclosporine A and mycophenolate mofetil were used in HSCT from haploidentical donors and mismatched unrelated donors after mid-2019. All patients had BM examination post-HSCT with documented morphological remission.
Definition
CR was defined by ⩽5% blasts in BM and complete haematologic recovery (absolute neutrophil count ⩾1 × 109/L and platelet count ⩾100 × 109/L). CR with incomplete haematologic recovery (CRi) was defined by ⩽5% blasts in BM, absolute neutrophil count <1 × 109/L or platelet count <100 × 109/L. The day of HSC infusion was defined as Day 0. Post-HSCT leukaemia-free survival (LFS) was defined as the duration from Day 0 to leukaemia relapse or death of any cause, with the data censored at last follow-up. Post-HSCT overall survival (OS) was defined as the duration between Day 0 and death of any cause, with the data censored at last follow-up. Cumulative incidence of relapse (CIR) was defined as the number of patients who relapsed, including morphological relapse (i.e. blasts ⩾5%) in BM, persistent circulating blasts in PB or extramedullary relapse at defined time points post-HSCT, with patients censored at the time of non-relapse mortality or at last follow-up. Clinical data were last updated on 3rd May 2024.
Mutation profile of patients
Targeted sequencing by NGS was performed on diagnostic (N = 198) and relapsed (N = 4) pre-HSCT samples, either in this study or by the referring hospitals. In this study, genomic DNA was extracted and NGS was performed using a pan-cancer panel comprising 544 recurrent genes or focused myeloid panels targeting 36–67 genes. Generation of DNA libraries, target capture, sequencing and bioinformatic procedures were described previously.5,7 In selected cases, genomic DNA was also extracted from BM samples at post-HSCT relapse for NGS using a focused myeloid panel targeting 36 genes. 5 Mutation profiles at diagnosis served to identify mutations amenable to MRD monitoring based on ddPCR or NGS. On the other hand, mutation profiles at haematological relapse tested the stability of specific mutations and justified their use as MRD markers.
MRD detection
BM aspiration was scheduled on Day 30 post-HSCT with adjustment based on clinical conditions and engraftment time. For patients with identified mutations amenable to MRD testing (Table S1), genomic DNA was extracted from the marrow blood and MRD detection was performed by ddPCR and NGS. Specifically, TaqMan digital PCR Mutation Detection Assay (Thermo Fisher Scientific, Waltham, MA, USA) was used for NPM1 Type A, IDH1 R132C and NRAS G12D mutation detection. Bio-Rad ddPCR Mutation Assay (Bio-Rad, Hercules, CA, USA) was used for DNMT3A R882H, IDH1 R132H/S, IDH2 R172K and FLT3 D835Y mutation detection. In-house ddPCR assays were designed for SF3B1 H662D/K700E, SRSF2 P95H, U2AF1 S34F and other patient-specific mutations, as described previously (Tables S1 and S3). 18 Bio-Rad QX200 Droplet Digital PCR System (Bio-Rad, Hercules, CA, USA) was used in these assays. Variant allele frequencies (VAF) were calculated by the number of mutant copies divided by the sum of mutant and wild-type (WT) copies. For targeted sequencing by NGS, the DNA sample (105 ng) was subjected to KAPA EvoPlus V2 enzymatic fragmentation, KAPA unique molecular identifier (UMI) adapter ligation, KAPA HyperCap hybridisation capture (Roche, Cape Town, South Africa) with 2X capture probe tiling density (IDT, Madison, WI, USA) and NovaSeq 6000 next-generation sequencing (Illumina, San Diego, CA, USA). UMI error-suppressed consensus sequencing reads were generated using Gencore version 0.17.2 19 and aligned to GRCh37/hg19 reference genome using BWA-MEM version 0.7.17. 20 BAM alignments of each called variant (mean consensus read depth 4000X) were manually reviewed and compared with unrelated WT samples in the same sequencing run using IGV version 2.18.1. 21 These WT samples served as the background signal. To define NGS MRD positivity, only those variants with the number of reads exceeding the background noise were recognised as true positives. When the background noise was very low, variants with 1 supporting consensus read could be accepted in the analysis.
Statistical analysis
Continuous data were compared using the Mann-Whitney U test. Categorical data were compared by Fisher’s exact test. Survivals were evaluated by Kaplan-Meier analysis and compared by log-rank test. p value <0.05 was considered statistically significant. CIR was evaluated using the competing risk approach and Gray’s test by tidycmprsk package version 1.1.0. 22 The median follow-up time was measured by Kaplan-Meier’s estimate as the median time to event (last follow-up), censored at death. The tests were conducted using Excel or R version 4.2.0. 23
To investigate the effects of different variables on survival outcome, univariate and multivariate analyses of clinical and genetic parameters, in addition to the MRD results, were analysed by the Cox-regression model in R using survival (version 3.7-0), 24 , survminer (version 0.5.0) 25 and tidycmprsk (version 1.1.0) 22 packages. The variables, including risk stratified by ELN 2022 classification, HSCT donor source, CR1 versus CR2 at HSCT, conditioning intensity, gene mutations with frequency of occurrence >15% and D30 MRD positivity, were included in the multivariate analyses 26 .
The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement. 22
Results
Patient cohort and overall treatment outcome
Among the 403 AML patients who received allo-HSCT at CR/CRi between 2003 and 2023 and had mutation information, 202 (50%) carried at least 1 of the 17 designated myeloid gene mutations (Table S1). MRD monitoring post-HSCT was performed in BM samples in 159 patients. Their clinicopathologic characteristics were shown in Table 1 and Figure 1(a). Median age at HSCT was 51 years (range: 20–67). A total of 112 (70%) and 45 (28%) patients received allo-HSCT at first and second remission respectively. Two other patients received HSCT at their third and fourth remission. The induction, consolidation and salvage regimens were summarised in Tables S4 and S5. HSC donors included human leukocyte antigen (HLA) identical (N = 70), matched (N = 40) and partially mismatched (N = 17) unrelated donors, and haploidentical donors (N = 32). Conditioning regimens were myeloablative in 103 (65%) and reduced intensity in 56 (35%) patients. Median time to neutrophil engraftment was 16 days (range: 10–31 days). Median follow-up time was 6.0 years (95% CI: 4.9–6.7). 41 (26%) patients relapsed after HSCT at a median of 10.8 months (range: 1.7–75.7). Median post-HSCT LFS and OS were not reached and 12.1 years respectively (Figure 1(b)–(d)). Fifty-one (32%) patients received some forms of intervention post-HSCT to prevent relapse, including treatment with FLT3 inhibitors (N = 19), hypomethylating agents (N = 35), venetoclax (N = 4) and donor lymphocyte infusion (N = 4) or their combinations (Table S6).
Clinicopathologic characteristics of the study cohort (N = 159).

CR, complete remission; CRi, CR with incomplete haematologic recovery; HSCT, haematopoietic stem cell transplantation.
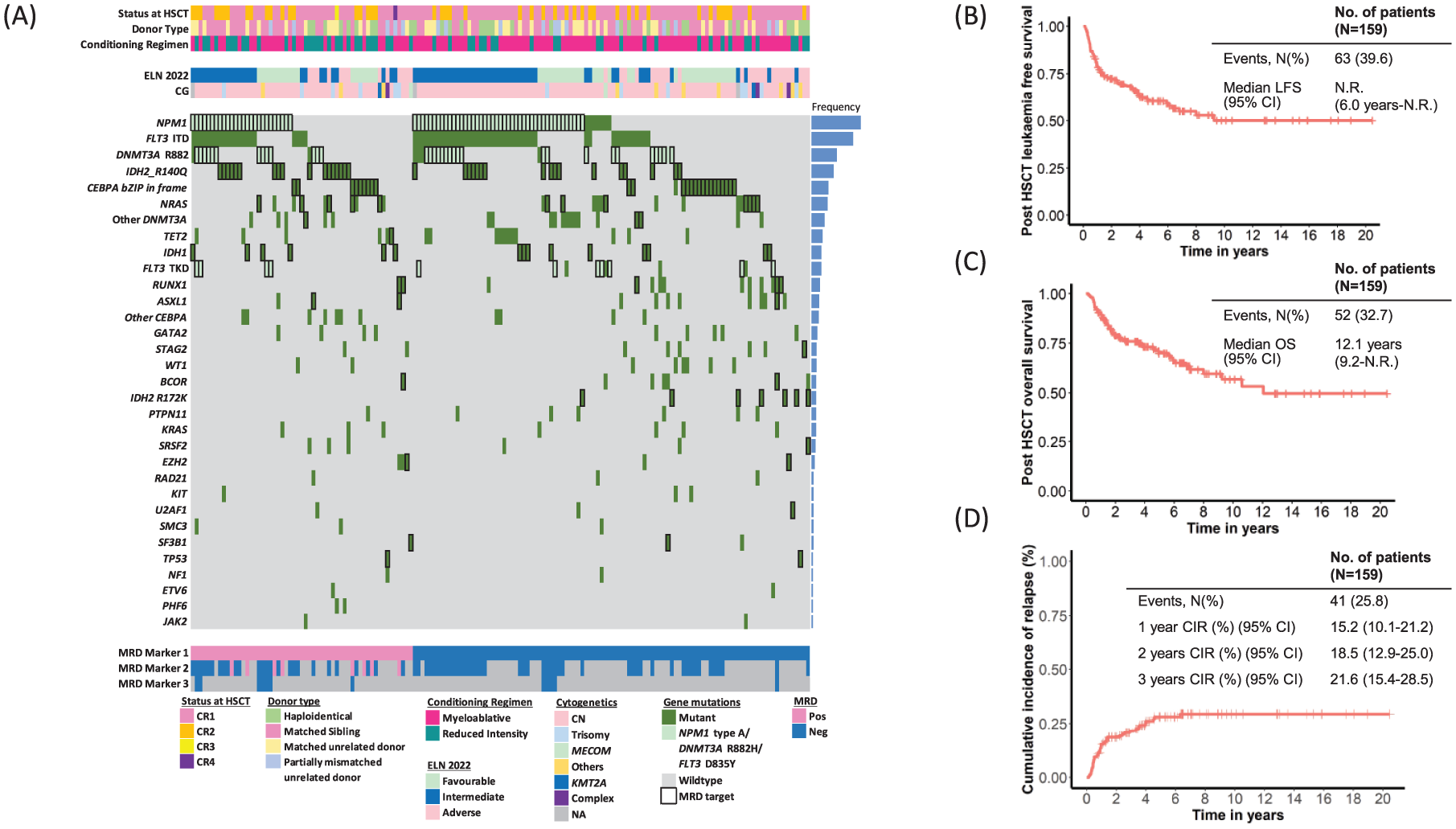
Clinicopathologic characteristics of the post-HSCT AML cohort in this study (N = 159). (a) Oncoplot showing the clinicopathologic characteristics, including disease status at which HSCT was performed, HSCT donor, conditioning regimen at HSCT, cytogenetics, ELN 2022 risk stratification, gene mutation status, MRD markers monitored in this study (thick border) and Day 30 MRD results. Each column represents a patient. Frequency of gene mutations in the cohort is shown as a bar chart on the right. (b) Post-HSCT LFS, (c) OS and (d) CIR of the cohort.
MRD detection
BM samples were available in 159 patients at a median of 26 days post-HSCT (range: 15–43 days). Patients with BM examination after 45 days were excluded. All showed morphological remission (blast <5%). In 122 patients, MRD detection was performed at nine recurrent mutations (NPM1 Type A, N = 70; DNMT3A R882H, N = 32; IDH1 R132H/C/S, N = 15; IDH2 R140Q/R172K, N = 41; FLT3 D835Y, N = 11; NRAS G12D, N = 12; SF3B1 H662D/K700E, N = 2; SRSF2 P95H, N = 1; and U2AF1 S34F, N = 1). In 38 patients, it was performed at nine genes with patient-specific mutations (CEBPA, TET2, TP53, ASXL1, BCOR, EZH2, RUNX1, STAG2 and DNMT3A non-R882 mutations; Figure 2(a); Table S1). The results were referred to hereafter as ‘D30 MRD’. Most MRD detection was based on ddPCR (N = 144), and NGS (N = 22) was mostly performed when the design of ddPCR was difficult, such as CEBPA mutations which were associated with high GC content at the mutation sites. MRD positivity was defined by cut-off values of VAF 0.001%–0.077% for ddPCR, depending on specific mutations and 0.1% for NGS-based testing (Table S1). In seven patients with CEBPA mutations where ddPCR design was possible, both ddPCR and NGS were performed, and the results showed high concordance (86%): five patients were MRD negative and one positive by both platforms. In one patient, NGS failed to identify a very low level of ddPCR-defined MRD positivity at 0.0031% VAF. In 78 patients, there was more than one MRD marker (two markers: N = 66; 3 markers: N = 12). Patients with any one MRD marker being positive were considered MRD positive.

Post-HSCT D30 MRD results and their association with LFS, OS and CIR. (a) Levels of D30 MRD in terms of variant allele frequency (%) among seven recurrent hotspots, CEBPA and other patient-specific mutations. Grey bars show the limit of sensitivity of each assay. Red circles represent the results from ddPCR, and blue circles those from NGS. Asterisks (*) denote that the limits of sensitivity of ddPCR assay and NGS range were between 0.00052%–0.01% and 0.05%–0.1% respectively for CEBPA bZIP in-frame mutation; 0.0066%–0.01% and 0.02%–0.1% respectively for other CEBPA mutations; 0.0011%–0.01% and 0.1% respectively for other patient-specific mutations. (b) Frequencies of D30 MRD positivity in patients receiving MAC and RIC during allo-HSCT. p Value of Fisher’s exact test is shown. (c–e) Kaplan-Meier curves showing the (c) LFS and (d) OS and (e) CIR between D30 MRD-positive and MRD-negative patients. p Values of log-rank test (c, d) and Gray’s test (e) are shown.
Despite morphological remission, D30 MRD was positive in 56 out of 159 patients (35%). MRD positivity was significantly associated with reduced intensity compared with MAC (p = 0.005 in Fisher’s exact test; Figure 2(b)) and in patients older than 50 years (p = 0.046 in Fisher’s exact test; Table S7). MRD positivity was not associated with specific gene mutations or ELN 2022 risk stratification (Tables S8 and S9).
Association of day 30 MRD with post-HSCT relapse
Among the 56 MRD-positive patients, 22 (39%) relapsed at a median time of 0.76 year. Among the 103 D30 MRD-negative patients, 19 (18%) relapsed at a median time of 1.1 years. D30 MRD positivity was associated with inferior post-HSCT LFS (median: 3.8 years vs not reached, p = 0.002) and OS (median: 10.6 years vs not reached, p = 0.019; Figure 2(c) and (d)). The CIR in MRD-positive and MRD-negative patients at 1 and 3 years post-HSCT was 26.9% versus 8.8% (p = 0.003), and 37.1% versus 13.2% (p = 0.0005) respectively (Figure 2(e)). A similar association between D30 MRD positivity and inferior LFS and higher CIR was observed within both subgroups of patients who received HSCT at CR1 and CR2. It was also associated with inferior OS in both subgroups, albeit statistically insignificant (Figure S1).
As the NPM1 mutation is the recommended MRD marker predictive of post-chemotherapy relapse, 27 its relevance in early post-HSCT was examined. D30 MRD positivity for NPM1 Type A mutation was associated with inferior LFS (median: 0.5 years vs not reached, p = 0.00012) and OS (median: 1.7 years vs not reached, p value = 0.0034). The CIR in NPM1 Type A mutation MRD-positive and MRD-negative cases at 1 and 3 years was 46.2% versus 6.8% (p = 0.0007) and 69.2% versus 16.9% (p = 0.00011) respectively (Figure 3(a)–(c)).

Post-HSCT LFS, OS and CIR in D30 MRD-positive and MRD-negative patients based on NPM1 Type A mutation (a–c) and mutations in non-NPM1 genes (d–f). Note that in panels (a)–(c), patients with negative NPM1 Type A D30 MRD but positive D30 MRD in non-NPM1 genes were excluded. Similarly, in panels (d)–(f), patients with negative D30 MRD in non-NPM1 genes but positive NPM1 Type A D30 MRD were excluded. p Values of log-rank test (a–b, d–e) and Gray’s test (c, f) are shown.
In addition, non-NPM1-mutated cases were examined. Their scarcity has precluded evaluation of individual mutations, and they were analysed together. D30 MRD positivity in non-NPM1 mutations was associated with a trend of inferior LFS (median 6.3 years vs not reached, p value = 0.064). The CIR in non-NPM1 mutation MRD-positive and MRD-negative cases at 1 and 3 years was 23.6% versus 10.4% (p value = 0.05) and 28.3% versus 11.8% (p value = 0.021) respectively (Figure 3(d)–(f)).
Factors associated with post-HSCT survival and post-HSCT MRD status
The effects of clinicopathologic characteristics, specific gene mutations, ELN 2022 risk stratification, disease status at HSCT, donor sources and conditioning, as well as D30 MRD status on post-HSCT LFS, CIR and OS were investigated. Among the various parameters, post-HSCT Day 30 MRD positivity was the only statistically significant factor associated with inferior post-HSCT LFS, CIR and OS in both univariate and multivariate analyses (Table S10 and Figure 4).

Multivariate analyses of clinicopathologic variables on post-HSCT (a) LFS, (b) OS and (c) CIR by Cox-regression model (a, b) and competing risks regression model (c). Hazard ratio with 95% confidence interval and p value was shown.
Assessment of MRD marker selection for post-HSCT MRD monitoring
To ascertain the stability of MRD markers at relapse, we performed NGS in nine relapsed samples, including five with positive and four with negative D30 MRD. Eight of them carried at least one of the mutations that were used as MRD markers, underscoring their stability at relapse and thus suitability for monitoring (Table S11). They included NPM1 Type A (N = 5), DNMT3A R882H (N = 2), IDH2 R140Q/R172K (N = 3), IDH1 R132H (N = 1) and FLT3 D835Y (N = 1). The remaining case was a patient (D30 MRD positive for NPM1 Type A and negative for DNMT3A R882H) who had an isolated relapse in the central nervous system 2.5 years post-HSCT. BM examination showed no blasts and no detectable mutations that were present at diagnosis.
Discussion
Post-HSCT leukaemia relapse is a major cause of treatment failure and mortality in the management of AML. While the European LeukemiaNet (ELN) 4 and National Comprehensive Cancer Network (NCCN) 28 have provided guidance on the use of NPM1 mutation and core-binding factor fusions as MRD monitoring tools during chemotherapy, the best timing of MRD detection post-HSCT and the role of other recurrent mutations in MRD have not been ascertained. Early post-HSCT flow or NGS-based MRD at 1 month were shown to predict leukaemia relapse, but the sensitivity was low compared to ddPCR assays.29,30 In this study, we hypothesised that MRD early post-HSCT could predict leukaemia relapse and reported that D30 MRD positivity, based primarily on highly sensitive ddPCR, is the single most important factor associated with shortened LFS and OS and increased relapse rate. In addition to the NPM1 mutation, MRD based on other recurrent mutations was of prognostic significance. Mutations chosen for MRD monitoring were stable at relapse in eight out of nine patients. Information arising from the present study not only confirmed findings from previous reports pertaining to the prognostic value of MRD post-HSCT,10,16,29,31–34 but might also shed new light on the management of AML.
First, we demonstrated the prognostic significance of mutations of NPM1 and non-NPM1 as MRD molecular markers post-HSCT. The results corroborated with recent reports on the prognostic relevance of NPM1 mutation MRD monitoring during early HSCT.34–36 NPM1 Type A mutation occurs in up to 42.6% of patients with cytogenetically normal AML 5 and is thought to be a driver in leukaemogenesis. The use of other recurrent myeloid mutations in MRD monitoring post-chemotherapy has raised concerns, as these mutations could reflect clonal haematopoiesis of indeterminate potential rather than a harbinger of leukaemia relapse. 17 In the post-HSCT setting, our results were consistent with previous reports that MRD based on non-NPM1 mutations is also of prognostic significance.29,33 The findings might broaden the application of MRD monitoring in other AML subtypes.
In addition, univariate and multivariate analyses consistently showed D30 MRD positivity as the single most significant factor predictive of outcome. MAC was associated with a significantly lower rate of D30 MRD positivity at the expense of higher transplant-related mortality, which might account for the lack of LFS or OS advantage in univariate and multivariate analyses.
Early detection of MRD positivity post-HSCT is of clinical relevance as it allows early enhancement of GVL effect via manipulation of the allo-immunity. The effects of donor leukocyte infusion (DLI) and early tapering of immunosuppressants are well recognised.4,37,38 More recently, hypomethylating agents4,37,39 and FLT3 inhibitors4,40–42 have also been shown to confer benefits post-HSCT by enhancing GVL effect. MRD-guided therapeutic intervention post-HSCT is beyond the scope of this study but should be systematically addressed. The differential impact of early intervention on HSCT outcome in different AML subtypes would have to be evaluated.
There are a number of limitations in this study. MRD was monitored mostly by ddPCR and, in some cases, NGS and was limited to mutations in the 17 myeloid-related genes for which molecular platforms have been established in our institute. The cohort in this study could not represent certain subtypes of AML, including those with chromosomal translocation as driver events or those not carrying these mutations. Moreover, different MRD platforms are needed for different mutations, and sometimes these platforms might show different sensitivities. This is in contrast to MFC, in which a single platform can be used, at the expense of a generally lower sensitivity compared with the ddPCR platform. Whether the latter could achieve better prognostication applicable to more AML subtypes post-HSCT would have to be evaluated. In addition, it is presently unclear if a multi-modality approach, including the combination of MFC, molecular platform and enrichment of HSPC, 43 which has been shown to increase detection sensitivity of MRD, would further improve the prognostic power of MRD. The sample size of the current study also limited the power to investigate the relative importance of individual genes used as MRD markers, the cut-off of MRD levels and the relevance of monitoring multiple MRD markers. Moreover, while the present study only focused on D30 MRD, the impact of pre- and late HSCT MRD and their temporal changes in the course of HSCT would have to be investigated. Previous studies have reported the prognostic impact of pre-HSCT MRD in predicting post-HSCT LFS and OS29,44–46 and the combination of MRD information in the peri-HSCT period might further improve the prognostic power.15,35 On the other hand, MRD at later time points, such as day 100, have also been shown to predict relapse.10,36 As BM assessment late in the course of HSCT is not a standard practice in our hospital, the number of samples was small, which such will limit the power of statistical evaluation. We reckoned that the detection of MRD early post-HSCT might provide an earlier window for intervention to prevent frank haematological relapse.
Conclusion
The present study demonstrated that D30 MRD positivity was predictive of a higher risk of relapse, and inferior LFS and OS post-HSCT. Early detection might allow intervention to prevent haematological relapse. The impact of post-HSCT MRD-guided intervention would have to be tested in the setting of a clinical trial.
Supplemental Material
sj-docx-1-tah-10.1177_20406207251389815 – Supplemental material for Early measurable residual disease detection post-haematopoietic stem cell transplantation in acute myeloid leukaemia
Supplemental material, sj-docx-1-tah-10.1177_20406207251389815 for Early measurable residual disease detection post-haematopoietic stem cell transplantation in acute myeloid leukaemia by Sze P. Tsui, Stephen S. Y. Lam, Chun H. Au, Chi Y. Fung, Beca B. K. Ip, Anthony T. C. Wong, Henry C. M. Leung, Wing H. Lai, Ho W. Ip, Edmond S. K. Ma, Garret M. K. Leung, Joycelyn P. Y. Sim and Anskar Y. H. Leung in Therapeutic Advances in Hematology
Supplemental Material
sj-docx-2-tah-10.1177_20406207251389815 – Supplemental material for Early measurable residual disease detection post-haematopoietic stem cell transplantation in acute myeloid leukaemia
Supplemental material, sj-docx-2-tah-10.1177_20406207251389815 for Early measurable residual disease detection post-haematopoietic stem cell transplantation in acute myeloid leukaemia by Sze P. Tsui, Stephen S. Y. Lam, Chun H. Au, Chi Y. Fung, Beca B. K. Ip, Anthony T. C. Wong, Henry C. M. Leung, Wing H. Lai, Ho W. Ip, Edmond S. K. Ma, Garret M. K. Leung, Joycelyn P. Y. Sim and Anskar Y. H. Leung in Therapeutic Advances in Hematology
Supplemental Material
sj-jpg-3-tah-10.1177_20406207251389815 – Supplemental material for Early measurable residual disease detection post-haematopoietic stem cell transplantation in acute myeloid leukaemia
Supplemental material, sj-jpg-3-tah-10.1177_20406207251389815 for Early measurable residual disease detection post-haematopoietic stem cell transplantation in acute myeloid leukaemia by Sze P. Tsui, Stephen S. Y. Lam, Chun H. Au, Chi Y. Fung, Beca B. K. Ip, Anthony T. C. Wong, Henry C. M. Leung, Wing H. Lai, Ho W. Ip, Edmond S. K. Ma, Garret M. K. Leung, Joycelyn P. Y. Sim and Anskar Y. H. Leung in Therapeutic Advances in Hematology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.