
Abstract
Background:
Early diagnosis of primary or light chain (AL) amyloidosis is crucial for initiating appropriate therapeutic interventions. However, diagnosis is getting delayed (several months-to-years) in clinical practice.
Objective:
To investigate the real-world patterns of clinical procedures until initial diagnosis of AL amyloidosis in Japan.
Design:
Retrospective longitudinal, observational cohort study.
Methods:
This study included adults with AL amyloidosis using Medical Data Vision claims database (2003–2022). The primary endpoint was time from initial hospital visit until confirmed AL amyloidosis diagnosis. Symptoms, lab tests, and medical department visits until diagnosis, and mortality were analyzed.
Results:
Overall, 323 patients with AL amyloidosis were included (median age: 73.0 years). Median time to confirmed diagnosis was 81.5 days; reported longer in patients aged ⩾65 years versus <65 years, and Charlson Comorbidity Index ⩾4 than <4. Specific tests (tissue and bone marrow biopsy) were conducted 28–40 days close to the diagnosis. Patients visited internal medicine (n = 158), hematology medicine (n = 139), dermatology (n = 97), and nephrology (n = 93) departments for confirmed diagnosis. Time to confirmed diagnosis was shorter for patients who visited hematology (median: 7.5 days). Early diagnosed (⩽1 year) patients had longer time-to-in-hospital death than late diagnosis (>1 year).
Conclusion:
These real-world data from Japanese AL amyloidosis patients are crucial for early and effective treatment, leading to better prognosis.
Keywords
Introduction
Primary or light chain (AL) amyloidosis, the most common form of systemic amyloidosis, is a rare protein misfolding and metabolism disorder characterized by the deposition of insoluble fibrils in various tissues, leading to impairment of organ function and death. 1 In 2018, global crude incidence of AL amyloidosis was 10.4 per million people with Japan having the highest incidence at 14.3 per million population. 2 Additionally, the prevalence of AL amyloidosis in Japan had steadily increased from 57.9 to 71.1 per million people from 2008 to 2018. 2 AL amyloidosis is associated with significant mortality with a 10-year relative survival rate of 27%. 2 AL amyloidosis presents with heterogeneous clinical manifestations, most commonly affecting the heart and kidneys, though the nervous, gastrointestinal, and hepatic systems may also be involved. 3
Despite significant progress in managing AL amyloidosis, including new combination therapies targeting novel pathways, diagnosis continues to be a challenge for clinicians across practice settings. 4 Patients are often misdiagnosed due to the nonspecific initial symptoms, including asthenia, dyspnea, heart failure with preserved ejection fraction, heart wall thickening, pseudo-infarction pattern, unexplained hepatomegaly, diarrhea, nondiabetic peripheral neuropathy, or nephrotic range proteinuria. 5 Additionally, hallmark clinical manifestations of AL amyloidosis, such as tongue enlargement or periorbital purpura, are reported in only 15% of patients. 5 The misdiagnosis and delay in diagnosis may lead to inadequate prognosis, high symptom burden, organ damage, and early deaths. 6
As per the Amyloidosis Research Committee of the Japanese Ministry of Health and Welfare, patients with AL amyloidosis are associated with more extensive organ involvement, necessitating increased awareness in early diagnosis and management of the disease. 7 Delayed diagnosis leads to disease progression, end-stage organ failure, and remains a major obstacle for initiating effective therapy. 8 Understanding the real-world clinical practice before diagnosis could help identify factors that contribute to diagnostic delays, supporting the early diagnosis and improving prognosis. Although the time to diagnosis of AL amyloidosis has been reported in several countries,9,10 real-world clinical practice leading to initial diagnosis in Japan remain unreported.
This retrospective study was designed to investigate the real-world patterns of clinical procedures leading to initial diagnosis of AL amyloidosis in Japanese patients. It focused on time to confirmed diagnosis, clinical lab tests, and visits to medical departments and hospitals, using data from an administrative claims database.
Material and methods
Study design and data source
This was a retrospective, longitudinal, observational cohort study, conducted in patients with AL amyloidosis using claims data from the Medical Data Vision Co., Ltd. (MDV, Tokyo, Japan) from 01 January 2003 to 31 December 2022. The MDV database is the largest healthcare database in Japan founded in 2003, comprising standardized administrative claims data from 480 hospitals in Japan (covering 28% of Diagnosis and Procedure Combination hospitals), providing insight on the diagnosis and treatment for nearly 48 million patients (as of July 2024).11,12 This database uses anonymized hospital-based medical data and claims from Japanese health insurance database, and enables analysis of data for rare diseases and diseases commonly seen in the elderly. 12 Given the retrospective nature and limited sample size of this real-world claim dataset, a primarily descriptive analytical approach was adopted to characterize diagnostic and clinical patterns without overfitting the data. The disease names documented in the MDV database are coded as per International Classification of Diseases, 10th Revision (ICD-10) and Japanese Disease Name Codes, and medical procedures are coded using Japanese Procedure Codes including medical departments. 13 Hospital deaths and other clinical evaluation data were obtained from the discharge summaries. As this analysis included extracted, de-identified, and anonymized patient data with secondary use, patient consent, and ethical approval were not required.
Study population
Adult patients aged ⩾18 years at the index date with a confirmed diagnosis record of AL amyloidosis (ICD-10 code, E85.81) between January 2003 and December 2022 were included. The index date was the first record of the confirmed diagnosis of AL amyloidosis between January 2003 and December 2022. The first confirmed diagnosis date should be within the period of the claims record available at the hospital.
Patients needed to fulfill any of the following criteria for inclusion in the study:
(i) First confirmed diagnosis record of AL amyloidosis after August 2021 or,
(ii) Record of combination therapy of melphalan and dexamethasone after the index date (Figure 1).
In August 2021, daratumumab was the first drug that received regulatory approval for treatment of AL amyloidosis in Japan. Since then, the combination therapy of daratumumab, cyclophosphamide, bortezomib, and dexamethasone was used, making diagnoses after August 2021 more reliable.

Patient identification flow in patients with AL amyloidosis.
Patients were excluded if they had,
(i) A hospital visit for the first time in the same month as the index date, and
(ii) Not received any treatment for AL amyloidosis (i.e., autologous peripheral blood stem cell transplantation, daratumumab, melphalan, bortezomib, lenalidomide, thalidomide, cyclophosphamide) within 1 year of confirmed diagnosis.
Study outcomes
The primary outcome of the study was to determine the time from initial visit to the hospital to the confirmed AL amyloidosis diagnosis. The time from initial visit of the hospital was defined as the evaluation period from the date of the first visit to the hospital which provided the confirmed diagnosis of AL amyloidosis to index date. Secondary outcomes included the patterns of clinical procedures until the initial diagnosis of AL amyloidosis with the following endpoints: time to the specific lab test (time from the initial visit of the hospital to the initial date of the specific lab test), time to specific medical department visit (time from the initial visit of the hospital to the initial date of the specific medical department visit), proportion of patients who took specific lab test and visited specific medical departments from the initial visit of the hospital, and the number and sequence of the lab tests and specific medical department visits the patients took from the initial visit to the hospital. Following AL amyloidosis diagnosis, time-to-in-hospitalized death was analyzed and compared between “Early diagnosed” and “Late diagnosed” groups. Early diagnosed group included patients who had ⩽1 year time from the suspected symptom to the confirmed AL amyloidosis diagnosis whereas ‘Late diagnosed’ group included patients who took >1 year time till diagnosis. Patients were followed up from initial hospital visits till the confirmed AL amyloidosis diagnosis. If the initial disease symptoms occurred before visiting the hospital, the initiation of evaluation period for the time to confirmed diagnosis was set as the first visit to hospital before the index date. Baseline demographics and clinical characteristics (age, gender, year of AL amyloidosis diagnosis, Charlson Comorbidity Index (CCI) score, comorbidities, etc.) were assessed. Tests classified as hematopoietic tumor antigen tests include flow cytometry or immunohistochemistry to detect plasma cell or hematologic malignancy markers (e.g., CD138).
Statistical analysis
Baseline demographics and clinical characteristics, primary and secondary endpoints except time-to-hospital death were summarized using descriptive analysis. Data were presented as number (n), mean (SD), median (min–max), frequency, and percentages. Kaplan–Meier analysis was used to present time-to-hospital deaths in patients with AL amyloidosis. The log-rank test was used to determine the probability of in-hospital deaths between the groups. All analyses were performed using SAS Version 9.4 (SAS Institute Inc., Cary, NC, USA) and R version 4.2.0.
We followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline in drafting this manuscript 14 and used the STROBE reporting checklist (refer Supplemental File) during the editing process. 15
Results
Baseline demographic and clinical characteristics
Overall, 13,491 patients’ data were retrieved from the MDV database. Following the eligibility criteria, 323 patients were included in the study (Figure 1). The mean ± SD age at index was 70.9 ± 10.0 years; 251 (77.7%) patients were aged ⩾65 years (Table 1). Overall, 186 (57.6%) patients were male, and 268 (83.0%) patients had an index date between 2016 and 2022. The mean ± SD CCI was 4.2 ± 2.6 years, and 184 (57.0%) patients had a CCI ⩾ 4. Among the baseline comorbidities, any malignancy including lymphoma and leukemia (n = 243, 75.2%), congestive heart failure (n = 185, 57.3%), renal disease (n = 97, 30.0%), peptic ulcer (n = 89, 27.6%), and mild liver disease (n = 70, 21.7%) were the most frequent comorbidities. Subgroup analyses were conducted by age group (<65 years vs ⩾65 years), CCI score (0–2 vs ⩾3), and timing of diagnosis (early vs late) to explore variations in diagnostic delays and treatment initiation.
Baseline demographics and clinical characteristics in patients with AL amyloidosis.

Data presented for >5% patients.
Except malignant neoplasm of skin.
AL, primary or light chain; CCI, Charlson Comorbidity Index; SD, standard deviation.
Time to the confirmed AL amyloidosis diagnosis
Overall, 228 patients with suspected diagnosis of AL amyloidosis at the initial visit were evaluated for time to confirmed diagnosis (Table 2). Median duration from the suspicion of symptoms to confirmed AL amyloidosis diagnosis was 81.5 days in the overall population. Median duration to confirmed diagnosis was longer in patients aged ⩾65 years than in patients aged 18–64 years (105.5 days vs 69.5 days). Patients who had a CCI ⩾ 4 had approximately 2–3 times longer median time to diagnosis than patients with a CCI of 0–3 (150.0 days vs 46.0–77.0 days). When stratified as per comorbidities (at least 10 cases), patients with chronic pulmonary disease had the longest (244.5 days) and those with any malignancy had the shortest (99.0 days) median time to diagnosis of AL amyloidosis.
Time from the suspected diagnosis initial visit of the hospital to the confirmed AL amyloidosis diagnosis (days).
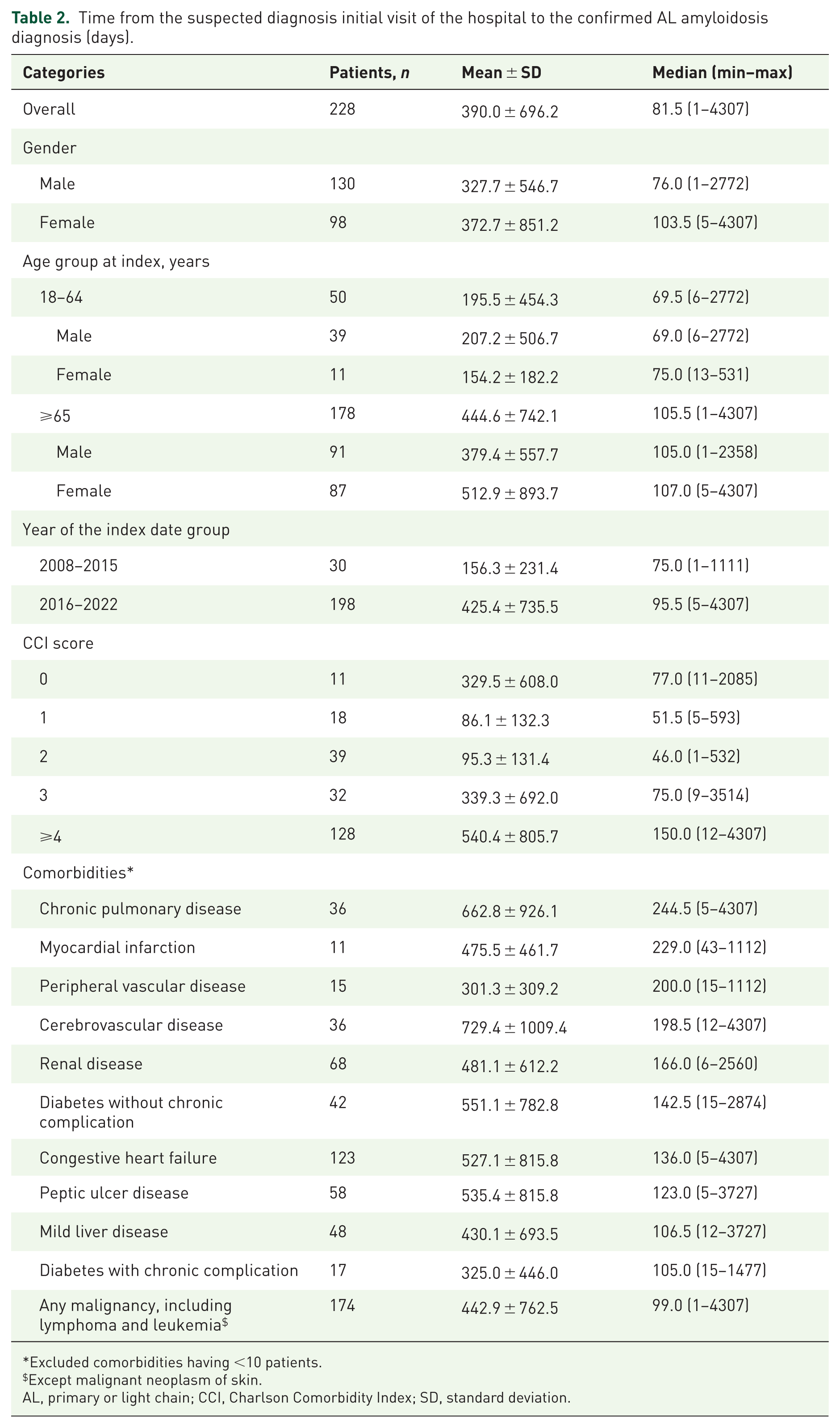
Excluded comorbidities having <10 patients.
Except malignant neoplasm of skin.
AL, primary or light chain; CCI, Charlson Comorbidity Index; SD, standard deviation.
Specific lab test to the confirmed AL amyloidosis diagnosis
All 323 patients underwent specific lab tests for the confirmation of the diagnosis. Specific lab tests such as tissue biopsy and bone marrow aspiration and myelogram were conducted shortly before the confirmed diagnosis (median, 28–40 days; Table S1). The median time from N-terminal pro b-type natriuretic peptide (NT-proBNP) testing (N = 243) to the confirmed diagnosis was 51 days (1–1422 days). The majority of patients received a confirmed diagnosis of AL amyloidosis after undergoing various tests, including bone marrow biopsy (n = 46), immunoglobulin free
Specific medical department visits to the confirmed AL amyloidosis diagnosis
All 323 patients had specific medical department visits for the confirmation of the diagnosis. A total of 29 medical departments were involved. Most of the patients visited internal medicine (n = 158), hematology medicine (n = 139), dermatology (n = 97), and nephrology (n = 93; Figure 2). Patients who visited the hematology department had the shortest median time (7.5 days) to confirm AL amyloidosis diagnosis (Table S2). Most patients who visited the internal medicine and hematology medicine departments confirmed AL diagnosis within 1 year (Table S3). Regarding the transition of medical department visits, patients had confirmed AL amyloidosis after transitioning to hematology (n = 64), internal medicine (n = 58), and dermatology (n = 52; Figure S2).

Number of patients who visited medical departments for confirmation of AL amyloidosis.
Confirmed prodromal symptoms in patients with AL amyloidosis
Most frequently reported confirmed prodromal symptoms in patients with confirmed AL amyloidosis diagnoses were heart failure (n = 151) and arrhythmia and cardiac blocks (n = 61) in cardiology and cardiopulmonary; nephrotic syndrome (n = 78) and chronic renal failure (n = 34) in renal; peripheral neuropathy (n = 33) and carpal tunnel syndrome (n = 5) in neurology; constipation (n = 94) and vomiting (n = 20) in GI/hepatic; and abdominal distention (n = 6) and glossitis (n = 3) in other therapeutic areas (Figure 3).
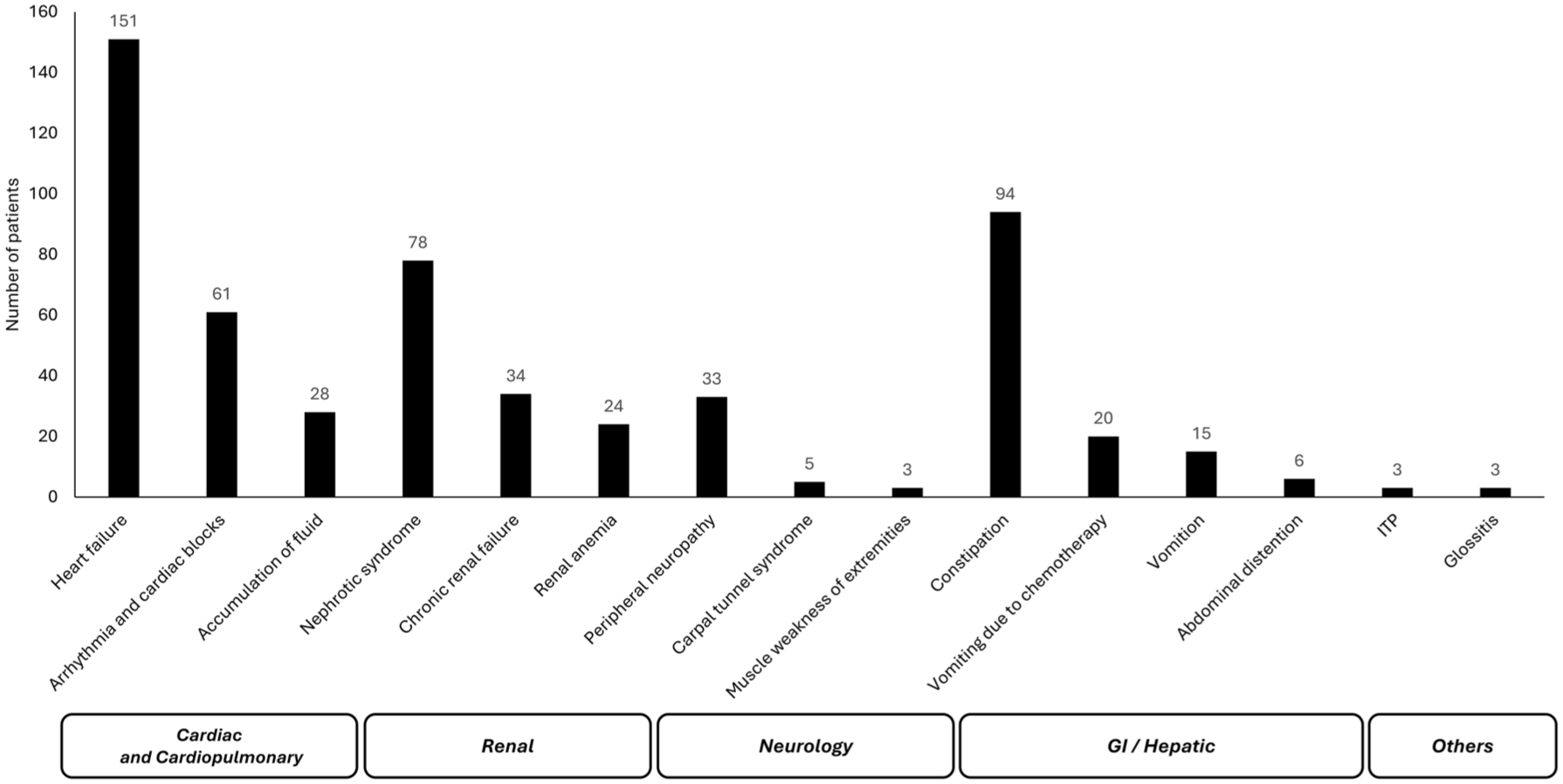
Number of patients with confirmed AL amyloidosis exhibiting clinical prodromal symptoms.
Time-to-in-hospital death after AL amyloidosis diagnosis
In total, 323 (early diagnosed, n = 225; late diagnosed, n = 78) patients with confirmed diagnosis and 228 (early diagnosed, n = 167; late diagnosed, n = 61) patients with suspected diagnosis were evaluated. The time-to-in-hospitalized death tended to be longer in the “Early diagnosed” group (time from suspected symptom to the confirmed AL amyloidosis diagnosis ⩽1 year) than in the “Late diagnosed” group (>1 year) in both confirmed (p = 0.905; Figure 4(a)) and suspected (p = 0.884; Figure 4(b)) AL amyloidosis diagnosis.

Time-to-in-hospitalized death after early versus late diagnosis of AL amyloidosis with (a) confirmed diagnosis, (b) suspected diagnosis. ‘Early diagnosed’ group: time from suspected symptom to the confirmed AL amyloidosis diagnosis ⩽1 year. ‘Late diagnosed’ group: time from suspected symptom to the confirmed AL amyloidosis diagnosis >1 year.
Discussion
This retrospective real-world observational study aimed to analyze the time from onset of suspected symptoms to confirmed diagnosis of AL amyloidosis, including associated clinical patterns, and in-hospital mortality following diagnosis, using the MDV database from 2003 to 2022. The extended time to diagnosis and frequent shifts between medical departments highlight the need for improved diagnostic strategies and increased physician awareness across specialties to facilitate earlier detection of AL amyloidosis.
The median duration from initial hospital visits for suspected diagnosis to confirmed AL amyloidosis diagnosis was approximately 3 months in our analysis. Moreover, patients aged ⩾65 years versus <65 years, and those with high CCI (>4 vs <4) had longer median time to confirm diagnosis. These time to diagnosis duration findings are mostly consistent or relative shorter that reported in the published literature.10,17 In contrast, some publications reported even longer time (2.7–3 years) from onset of symptoms to confirmed diagnosis of AL amyloidosis.9,18
In the current analysis, median age of the overall cohort was 73.0 years with >75% patients aged ⩾65 years, and minor male predominance (57.6%). This finding is aligned with the existing evidence that AL amyloidosis predominantly affects older adults, with males being slightly more affected than females.3,7,9,19,20 Moreover, patients with AL amyloidosis consistently had high CCI, and were primarily suffering from cardiac- and renal-related diseases3,7,9,10,19 as observed in our analysis. Additionally, older patients and patients with comorbidities such as chronic pulmonary disease, myocardial infractions, peripheral vascular disease, and cerebrovascular disease had longer time to confirmed diagnosis of AL amyloidosis in our analysis. This might be attributed to the presence of nonspecific or similar symptoms in patients with AL amyloidosis that creates the delay in confirmed diagnosis. 21
Consistent with the guideline and published literature,4,10,22,23 in our analysis, patients underwent specific lab tests and immunochemistry-specific tests for the confirmation of the diagnosis. The biopsy was done in close timing to the confirmed diagnosis of AL amyloidosis. In certain cases, physicians preliminarily consider the diagnosis as malignancy and accelerate the specific tests such as biopsy, leading to early diagnosis of AL amyloidosis. Usually, the test results of any disease can be obtained in 1–2 months of diagnosis. 18 However, it takes nearly a year in certain cases, which may be due to the quality of biopsy/tissue results that needs a re-biopsy, hospitals being unfamiliar with the test, frequency of patients’ hospital visit, or physicians don’t have enough time to see the test results. 3 Additionally, the uncertainty of immunostaining for determining the type of AL amyloidosis could add additional challenge in accurate diagnosis of the disease. 24 Also, potentially there are cases where biopsy and confirmed diagnosis were accelerated because physicians’ prioritized diagnosing malignancy. These parameters highlight the need for disease awareness among patients and physicians for early diagnosis and informed treatment decision making.
As AL amyloidosis is associated with nonspecific symptoms, patients need to visit several clinicians and medical departments to confirm the diagnosis. Among 29 medical departments involved in our study, patients were primarily visited to the internal medicine, hematology, dermatology, and nephrology departments for the confirmation of AL amyloidosis diagnosis. Moreover, patients were mostly transitioned from the hematology, internal medicine, and dermatology departments for the confirmed diagnosis. A retrospective database study including Japanese patients with AL amyloidosis reported similar findings as referrals were mostly from the hematology, nephrology, cardiology, and internal medicine departments for the diagnosis confirmation. 3 Nonetheless, an integrated approach combining inputs from different clinicians with expertise in amyloidosis could facilitate in the accurate diagnosis and specialized treatment of AL amyloidosis. 23
Cardiac and renal involvement, especially heart failure and renal disease, are the initial manifestations of AL amyloidosis.3,19,20 Moreover, studies also reported neurologic and GI manifestations in some cases.9,18 Consistent with these findings, our analysis identified heart failure, constipation, nephrotic syndrome, arrhythmia and cardiac blocks, peripheral nephropathy, and chronic renal failure as the most frequent confirmed prodromal symptoms in patients with confirmed AL amyloidosis. These overlapping manifestations make it challenging for clinicians to accurately diagnose AL amyloidosis, often leading to delays and multiple medical department visits before a confirmed diagnosis.
As a progressive, multiorgan disease, delays in diagnosing AL amyloidosis can elevate the risk of mortality due to associated comorbidities.19,20,25,26 Cardiac, renal, and hepatic involvement are the most prominent causes of increased mortality in AL amyloidosis.10,22,25 Approximately one-third of patients with AL amyloidosis with cardiac involvement experienced early death within 5 months of diagnosis, highlighting the importance of early disease identification. 10 In our analysis, patients who were diagnosed early (⩽1 year from suspected symptoms to confirmed diagnosis) had longer time-to-in-hospital mortality than patients who diagnosed late (>1 year). A retrospective observational study from Germany reported that patients who diagnosed AL amyloidosis after 1 year of first symptoms had a 3–5 times higher risk of mortality compared with patients who diagnosed within the first year. 26 These findings highlight the critical importance of early diagnosis and support timely therapeutic interventions to improve patient outcomes. Although this is a population-based study representing population of Japan and provides a real-world scenario in the field of AL amyloidosis, the findings should be interpreted with caution in global population. Our findings, though based on Japanese claims data, align with international reports of diagnostic delay and clinical complexity in AL amyloidosis. These diagnostic challenges are not unique to Japan and suggest a need for greater awareness and early screening protocols globally.
Limitations
Since MDV database is the largest database that includes information about all diseases and procedures, it only captures the hospitalized data. In our analysis, the data regarding patient death was only confined to hospitalized patients. Sample size calculation was not performed as this was a retrospective analysis using preexisting claims data; this limits the ability to assess the statistical power for specific outcomes. Moreover, the study was limited by a small sample size, retrospective nature, and the possibility that database-specific claims for AL amyloidosis could be under-reported in database studies. 20
Due to the modest sample size and data constraints, multivariate statistical modeling was not feasible. Future research using larger datasets or clinical registries should consider multivariable approaches to adjust for potential confounders. Additionally, our analysis was limited to patients with confirmed AL amyloidosis and predominantly descriptive. Hence, time to diagnosis could be shorter when compared to the clinical setting as there might be many patients who would die before getting confirmed diagnosis or might be completely misdiagnosed or unable to receive appropriate treatment. As our study did not exclude patients with concurrent multiple myeloma, some cases with both conditions may have been included, which reflects the real-world spectrum of AL amyloidosis presentation. Finally, >75% of patients had malignancy at baseline in our study. This might not reflect actual comorbidity as the information was collected from a database. Nonetheless, this study could provide useful data on factors that could delay the diagnosis of AL amyloidosis to inform personalized management strategies.
Conclusion
This study offers insights into diagnostic patterns in Japanese patients with AL amyloidosis, including the time to diagnosis and clinical patterns involved prior to confirmed diagnosis, and also highlights the importance of early diagnosis in reducing patient mortality. These data increased awareness regarding early diagnosis and emphasizes the importance of specific and quick laboratory tests, medical department visits to improve the outcomes of patients with AL amyloidosis with reduction in early mortality, and effective treatment. Future studies are needed to explore the comorbidity-specific mortality, risk factors for extended duration of diagnosis using real-world data, treatment pattern based on organs involved, and long-term treatment outcomes in patients with AL amyloidosis.
Supplemental Material
sj-docx-3-tah-10.1177_20406207251379852 – Supplemental material for A retrospective real-world study assessing diagnostic pattern of light-chain amyloidosis in Japan based on data from the medical data vision claims database
Supplemental material, sj-docx-3-tah-10.1177_20406207251379852 for A retrospective real-world study assessing diagnostic pattern of light-chain amyloidosis in Japan based on data from the medical data vision claims database by Moe Yogo, Mami Kasahara-Kiritani, Kazuki Oshima and Tadao Ishida in Therapeutic Advances in Hematology
Supplemental Material
sj-jpg-1-tah-10.1177_20406207251379852 – Supplemental material for A retrospective real-world study assessing diagnostic pattern of light-chain amyloidosis in Japan based on data from the medical data vision claims database
Supplemental material, sj-jpg-1-tah-10.1177_20406207251379852 for A retrospective real-world study assessing diagnostic pattern of light-chain amyloidosis in Japan based on data from the medical data vision claims database by Moe Yogo, Mami Kasahara-Kiritani, Kazuki Oshima and Tadao Ishida in Therapeutic Advances in Hematology
Supplemental Material
sj-jpg-2-tah-10.1177_20406207251379852 – Supplemental material for A retrospective real-world study assessing diagnostic pattern of light-chain amyloidosis in Japan based on data from the medical data vision claims database
Supplemental material, sj-jpg-2-tah-10.1177_20406207251379852 for A retrospective real-world study assessing diagnostic pattern of light-chain amyloidosis in Japan based on data from the medical data vision claims database by Moe Yogo, Mami Kasahara-Kiritani, Kazuki Oshima and Tadao Ishida in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
The authors thank Rabi Panigrahy, PhD (SIRO Clinpharm Pvt Ltd) for writing assistance and Shweta Pitre, MPH, CMPP™ (SIRO Clinpharm UK Ltd) for additional editorial support for the development of the manuscript, which was funded by Janssen Pharmaceutical K.K., Japan.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.