
Abstract
Waldenström macroglobulinemia is an indolent B-cell lymphoma which although remains incurable, there are a lot of treatment options. Today, Bruton tyrosine kinase inhibitors have a central role in the management of the disease either as monotherapy or combination with other regimens, due to their efficacy, ease of administration, and safety profile. However, there is still active clinical investigation to further increase their efficacy and improve safety profile. Combinations based on BTK inhibitors may offer advantages. Second- and third-generation BTK inhibitors are also evaluated in combinations aiming to improve the depth of response, overcome genetic factors associated with poorer outcomes and reduce toxicity and duration of therapy.
Keywords
Introduction
Waldenström’s macroglobulinemia (WM) is an indolent B-cell lymphoma characterized by the infiltration of the bone marrow by lymphoplasmacytic cells that produce various amounts of monoclonal immunoglobulin M (IgM). 1 WM is a disease of the elderly with a median age at the time of diagnosis being around 70–71 years. 2 It is a rare disease (2% of all lymphomas), with a 2:1 male predominance and higher incidence among Caucasians.3–5 Interestingly, a familial predisposition has also been described.6,7 Genetically, WM is characterized by the presence of clonal MYD88 mutations that are observed in >90% of patients, while in up to 40% somatic CXCR4 mutations are found, and which are associated with higher bone marrow burden, serum IgM levels and risk of hyperviscosity, shorter time from asymptomatic to symptomatic WM, and relative resistance and shorter duration of response (DOR) to some treatments.5,8
Our therapeutic armamentarium to manage WM includes a variety of options with favorable results9–11; however, it still remains an incurable disease, following a natural history that is characterized by remissions and relapses requiring management with different therapies. The introduction of Bruton tyrosine kinase (BTK) inhibitors offered the most effective single-agent therapy for WM but also introduced new challenges, since these drugs are offering unparalleled response rates but cannot eliminate and require continuous therapy to control the disease. In addition, the genetic profile of the clone (the presence of MYD88L265P and of mutations in CXCR4) can affect the efficacy of BTKs, introducing the need for combination therapies to overcome these challenges. The aim of this review is to present the current data on BTK inhibitors (BTKs) as single agents in frontline and previously treated patients or as combinations with other regimens and elucidate their role as part of the sequential therapy in WM.
BTKs inhibitors in WM
BTK inhibitors play a significant role in the management of B-cell malignancies targeting B-cell receptor (BCR) signaling pathways. Ibrutinib is the first-in-class BTK inhibitor which forms an irreversible covalent bond with cysteine residue at position 481, leading to a constant inhibition of BTK even after the metabolism of the drug.12,13 Ibrutinib is the first drug that received approval for WM in 2015 by the Food and Drug Administration (FDA). 13 It is administered orally with a recommended dose of 420 mg daily. However, ibrutinib is a moderately selective BTK inhibitor that also inhibits other off-target cysteine-containing kinases which may result in adverse events. 12 The most common toxicities of ibrutinib are bleeding diathesis, cytopenias, infections, and atrial fibrillation. 13 Moreover, the presence of somatic CXCR4 mutations limits the efficacy of ibrutinib. 14 Second-generation covalent BTK inhibitors, such as zanubrutinib, acalabrutinib, tirabrutinib, and orelabrutinib, have been developed. Zanubrutinib gained approval by the FDA for WM in 2021. These second-generation BTKs also form covalent, irreversible bonds with the cysteine 481 residue, but with higher selectivity. 12 Zanubrutinib is associated with significantly lower risk for bleeding and atrial arrhythmias but higher risk of cytopenias than ibrutinib in a head-to-head comparison. 15 Although acalabrutinib is not yet approved for WM, it is characterized by higher selectivity with somewhat improved toxicity profile compared to ibrutinib 16 ; mostly regarding lower rates of atrial arrhythmias 17 and lower frequency of hemorrhagic complications. 18
Despite the use of second-generation covalent BTK inhibitors that have a more favorable profile concerning cardiovascular toxicity, this remains significant. The most common cardiovascular complications of covalent BTK inhibitors that have been reported include hypertension, atrial fibrillation, and ventricular arrhythmias, including sudden cardiac deaths. 19 Although second-generation BTK inhibitors are safer, with significantly lower risk of arrhythmias and hypertension, the decision to use covalent BTK inhibitors should be personalized based on their toxicity profile and the cardiovascular co-morbidities of the patient. 15 Third-generation (noncovalent) BTK inhibitors, such as pirtobrutinib and nemtabrutinib, have also been investigated in WM. These are reversible BTK inhibitors, with pirtobrutinib being more selective than nemtabrutinib. 12 Noncovalent BTK inhibitors have shown efficacy even in covalent BTK inhibitor resistant disease and a safer toxicity profile, especially concerning cardiovascular complications.20–22
Acquired, and (less commonly) inherent, resistance to BTK inhibitors is a significant therapeutic challenge. Mechanisms of resistance to BTK inhibitors have been more extensively studied not only in chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), diffuse large B-cell lymphoma but also in patients with WM. BTK inhibitors block BTK signaling through binding to Cys-481 thus leading to the inhibition of the BCR pathway. Amino acid substitutions of Cys-481 (replacement of cysteine with other amino acids, such as serine (C481S) or arginine (C481R)) of BTK or mutations in the phospholipase C gamma 2 gene (PLCG2) are some common acquired mechanisms of resistance to BTK inhibitors. 23 Continuous use of covalent BTK inhibitors can lead to acquired mutations resulting in activation of ERK1/2 pathway and subsequently resistance to the site of bond with cysteine 481 residue.22,24 Moreover, 8p loss is another acquired resistant mechanism to covalent BTK inhibitors. 23 This deletion has been noticed in patients refractory to BTK inhibitors without mutations of BTK or PLCG genes.23,25 In WM, deletions on chromosomes 6q (including homozygous deletions) and 8p, recurring mutations in ubiquitin ligases, innate immune signaling, and TLR/MYD88 pathway regulators have also been implicated in resistance to BTKs (mainly ibrutinib). 25 There is also data supporting that patients with CXCR4 mutations are prone to acquired resistance to BTK inhibition. 24 The surface receptor CXCR4 interacts with the chemokine CXCL12 resulting in the activation of Akt signaling which leads to prolonged survival of the cancer cells. 23 Noncovalent BTK inhibitors such as pirtobrutinib seem to provide a solution to this limitation, as they can overcome resistance related to C481S mutations. In the BRUIN study, patients refractory to ibrutinib responded to pirtobrutinib, but the rates, depth, and duration of these responses were less optimal than in non-cBTK resistant patients. Data from the CLL field indicate that other second-site acquired BTK mutations may lead to resistance to pirtobrutinib. Thus, additional clinical trials are needed to clarify the exact place of noncovalent BTK inhibitors in the sequence of treatments for WM and the mechanisms of resistance and clonal escape. 14
Monotherapy with BTK inhibitors
Ibrutinib
Ibrutinib has been investigated either as a single agent or in combination with other regimens in the frontline of WM or in previously treated patients. The results of these clinical trials are described in Tables 1 and 2. The pivotal study of ibrutinib monotherapy that led to its approval included 63 relapsed/refractory WM patients without prior exposure to ibrutinib. 26 The highest response rates were recorded among patients with MYD88L265P and CXCR4 wild-type genotypes, being the first study that demonstrated the impact of MYD88 and CXCR4 mutation status in treatment response. The long-term results of the study confirmed the high response rates (overall response rate (ORR) of 90.5% and major response rate (MRR) of 79.4%), and a favorable 5-year progression-free survival (PFS) and overall survival (OS) of 54% and 87%, respectively. 27
Ibrutinib in WM.

Referred to both TN and RR pts.
Twelve patients were analyzed.
AE, adverse events; CR, complete response; f/up, follow-up; Gr, grade; I, ibrutinib; m, median; mo, months; mOS, median overall response; mPFS, median progression-free survival; MR, minor response; MRR, major response rates; n, number; No, number; ORR, overall response rate; OS, overall response; PD, progressive disease; PFS, progression-free survival; PR, partial response; pts, patients; R, rituximab; RR, relapsed/refractory; SD, stable disease; TN, treatment naïve; VGPR, very good partial response; WM, Waldenström macroglobulinemia.
Other clinical trials of ibrutinib in WM.
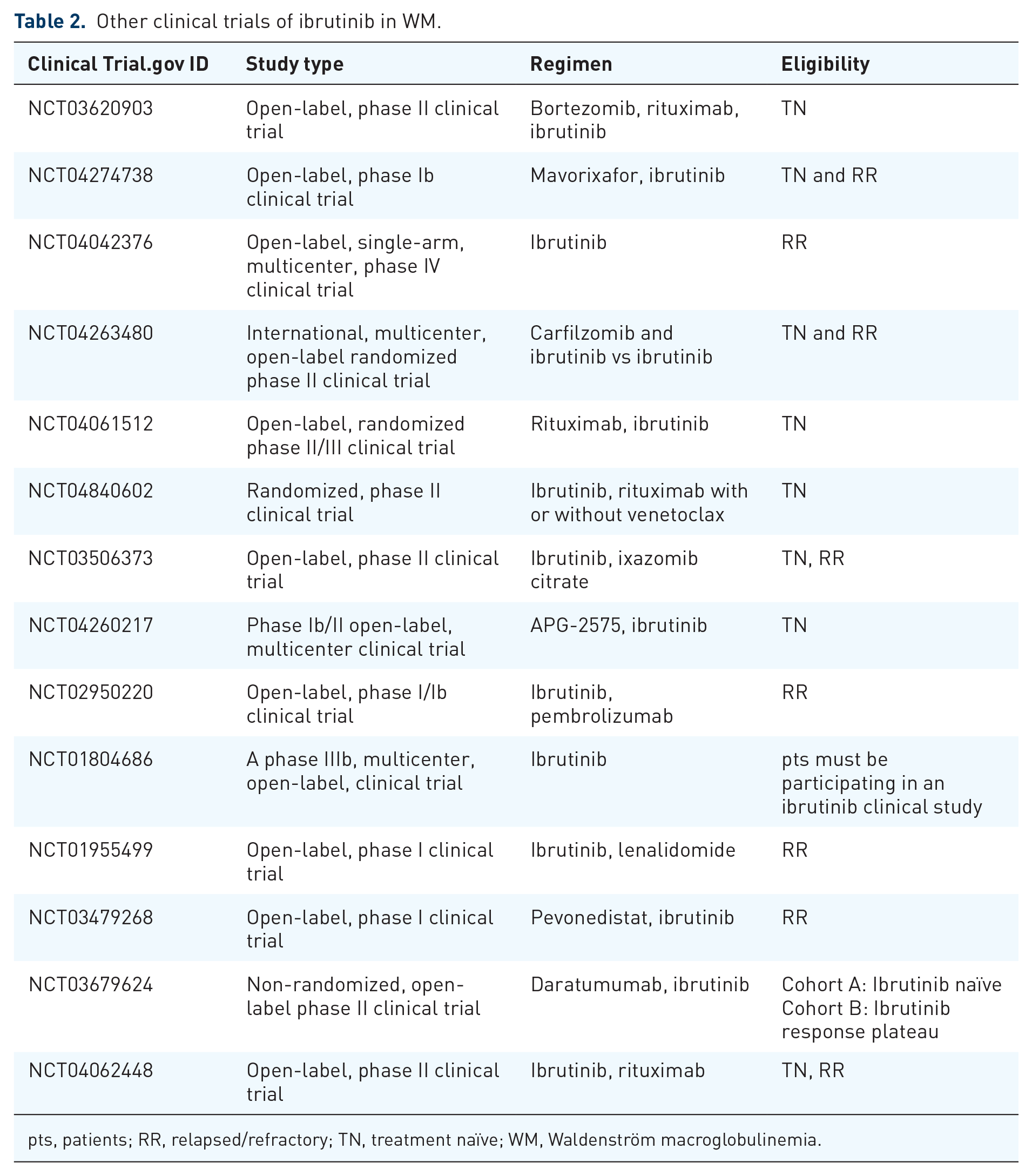
pts, patients; RR, relapsed/refractory; TN, treatment naïve; WM, Waldenström macroglobulinemia.
In a substudy of the iNNOVATE trial, ibrutinib was evaluated as a single agent in 31 pretreated WM patients who were rituximab-refractory. 28 At a median follow-up of 50 months, the ORR and MRR were 87% and 77.4%, respectively, while the median PFS was 39 months and 5-years PFS was 40%. 29 Notably, in patients with CXCR4 mutations the 60 months PFS was 60% versus 0% (median 18 months). Hence, ibrutinib monotherapy in WM patients with disease that is refractory to rituximab-based therapy is an option with significant efficacy and durable responses, especially when CXCR4 mutations are absent.
Ibrutinib monotherapy in previously untreated symptomatic patients with WM also demonstrated favorable results.30,31 In a study that enrolled only patients harboring MYD88L265P, after a median follow-up of 50 months, ORR and MRR were 100% and 87%, with a 4-year PFS rate of 76%. Consistent with findings in the pivotal study of ibrutinib, the presence of CXCR4 mutations unfavorably affected response rates and PFS. 26
Abeykoon et al. 32 presented real-world (retrospective) evidence concerning the efficacy and safety of ibrutinib monotherapy in both treatment-naïve and previously treated WM patients. The MRR was 78% and the 18-month PFS was 82%. However, the researchers underlined that the use of ibrutinib has challenges as 31% of the enrolled patients discontinued ibrutinib mainly due to toxicities and 18% required a decreased dose. Acquired resistance to ibrutinib monotherapy is an important challenge. Combinations of bendamustine–rituximab (BR) and bortezomib–dexamethasone–rituximab (BDR) have shown activity, especially in patients without prior exposure to them.33,34 Outside the context of clinical trials, Abeykoon et al. 32 provided data that after ibrutinib discontinuation chemoimmunotherapy regimens such as BR or dexamethasone–rituximab–cyclophosphamide (DRC) were effective as salvage options. Proteasome inhibitor-based regimens may be another options for such patients while the introduction of noncovalent BTK inhibitors (such as nemtabrutinib and pirtobrutinib) and of BCL-2 inhibitors (such as venetoclax or sonrotoclax) which have also shown activity in ibrutinib refractory or intolerant patients may be other options and will be discussed in more detail.
Zanubrutinib
Zanubrutinib is a selective, irreversible second-generation covalent BTKi 35 ; this drug has been investigated in several clinical trials in WM either as monotherapy or in combinations as described in Table 3.
Zanubritinib in WM.

Forty-three patients were analyzed.
Including all patients with B-cell malignancies.
Referred to 64 efficacy-evaluable patients.
Referred to both TN and RR pts.
AE, adverse events; CR, complete response; DOMR, duration of major response; DOR, duration of response; f/up, follow-up; Gr, grade; m, median; mDOR, median duration of response; mo, month; mOS, median overall survival; mPFS, median progression-free survival; MR, minimal response; MRR, major response rate; n, number; NA, not available; No, number; OR, overall response; ORR, overall response rate; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PR, partial response; pts, patients; RR, relapsed/refractory; SD, stable disease; TN, treatment naïve; VGPR, very good partial response; WM, Waldenström macroglobulinemia.
Zanubrutinib has been administered as monotherapy either in frontline or in relapsed/refractory WM patients achieving deep (but still not complete) and durable responses.36,37 The first studies in WM, by Trotman et al., 36 showed very high response rates among treatment-naïve as well as in pretreated patients (ORR: 100% vs 93.9%, PFS: 91.5% vs 76.2%).
In the ASPEN trial, the safety and efficacy of zanubrutinib was compared directly to that of ibrutinib.15,38 The primary endpoint of the study was the rates of CR and very good partial response (VGPR): this was not met, although they were numerically higher with zanubrutinib (28% and 19%), and there were no CRs. ORR and MRR were similar (93.9% vs 95.1% and 79.8% vs 81.4%, respectively) with the 42-month event-free PFS and OS being 69.7% and 85.2% for ibrutinib versus 78.3% and 87.5% for zanubrutinib, respectively. Importantly, higher rates of VGPR were recorded on zanubrutinib-treated patients carrying CXCR4 mutations compared to ibrutinib arm (21.2% vs 10%) as well as in treatment-naïve and CXCR4WT patients. In a subanalysis from this study, zanubrutinib had a more favorable activity profile over ibrutinib in genetic groups such as patients with CXCR4 non-sense (NS) mutations and those harboring TP53 mutations. In an ASPEN substudy (cohort C), zanubrutinib monotherapy was given in 28 patients (23 relapsed or refractory) with wild-type MYD88. The MRR was 50% and 18-month PFS and OS were 68% and 88%, respectively. In this direct comparison, the toxicity profile of zanubrutinib was more favorable than that of ibrutinib, and in combination with the higher efficacy in certain genotype groups, provides evidence to support its use over ibrutinib. Although data are limited about the sequence of covalent BTKis, in a multicenter study, patients intolerant to ibrutinib or acalabrutinib received zanubrutinib. 39 The results revealed that among 64 evaluable patients the ORR was 64.1%, suggesting that zanubrutinib could be an option for those patients that are intolerant to ibrutinib or acalabrutinib, being a better tolerated treatment.
Other ongoing clinical trials examining zanubrutinib, either as monotherapy in previously untreated patients or as combination with other regimes in pretreated or naïve treatment patients, are presented in Table 4.
Other clinical trials of zanubrutinib in WM.

pts, patients; RR, relapsed/refractory; TN, treatment naïve; WM, Waldenström macroglobulinemia.
Acalabrutinib in WM
Acalabrutinib is a highly selective, irreversible covalent BTKi that has been approved for the treatment of MCL and CLL/small lymphocytic lymphoma (CLL/SLL).40,41 Although it has not been approved for WM, it has been investigated either as a monotherapy or in combination with other regimens, as shown in Tables 5 and 6.
Acalabrutinib in WM.

CR, complete response; Gr, grade; f/up, follow-up; mo, months; mOS, median overall survival; mPFS, median progression-free survival; MRR, major response rate; n, number; NA, not available; No, number; ORR, overall response rate; OS, overall survival; PR, partial response; pts, patients; RR, relapsed/refractory; TN, treatment naïve; VGPR, very good partial response; WM, Waldenström macroglobulinemia.
Other clinical trials of acalabrutinb in WM.

pts, patients; TN, treatment naïve; WM, Waldenström macroglobulinemia.
In a single-arm, multicenter phase II study, single-agent acalabrutinib was evaluated in treatment-naïve and relapsed/refractory patients with WM.42,43 Responses rates were high, with ORR and MRR similar among pretreated and untreated patients, although the estimated 66-month DOR and PFS were higher in treatment-naïve patients (90% vs 45% and 84% vs 52%, respectively). In this study, there was no systematic evaluation of the effect of genotype to the efficacy and DOR after acalabrutinib, but a post hoc analysis in a subgroup of patients with available data indicated higher MRR among MYD88L265P versus MYD88 wild-type patients.
Other covalent BTK inhibitors
Other covalent BTK inhibitors that are investigated in WM are tirabrutinib and orelabrutinib. Tirabrutinib is an oral, irreversible, covalent BTK inhibitor that was approved in Japan in 2020 for recurrent or refractory primary central nervous system lymphoma. 44 Orelabrutinib is a novel, covalent, irreversible BTK inhibitor, orally administered, with high selectivity. 45 It has been previously approved in China for relapsed/refractory patients with MCL and CLL/SLL. 46 Both of these two BTK inhibitors have been evaluated as monotherapy in WM with high responses rates as shown in Table 747–51: with tirabrutinib the ORR and PFS at 2 years were 96.3% and 92.6%, respectively, 50 and with orelabrutinib an ORR of 90.3% was achieved at 12 months and the estimated 1 year PFS was 89.4%. 51 These BTK inhibitors may be associated with a lower risk of atrial arrhythmias or bleeding compared to ibrutinib, and they seem to have similar efficacy.
Other covalent BTK inhibitors in WM.

Referred to both TN and RR patients.
BTK, Bruton tyrosine kinase; f/up, follow-up; Gr, grade; mo, months; mOS, median overall survival; mPFS, median progression-free survival; MRR, major response rate; n, number; NA, not available; No, number; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; PR, partial response; pts, patients; RR, relapsed/refractory; TN, treatment naïve; VGPR, very good partial response; WM, Waldenström macroglobulinemia.
Noncovalent BTK inhibitors
Noncovalent BTK inhibitors block BTK signaling by different mechanisms than covalent BTKis. They do not bind to the C481 residue of BTK, and thus, can overcome one of the mechanisms of resistance to covalent BTKs (i.e., mutations in the C481). In addition, due to their higher selectivity and reversible binding to BTK they have a more favorable toxicity profile. Pirtobrutinib (previously known as LOXO-305) is a reversible, noncovalent BTK inhibitor with evidence of activity in WM cells that harbor BTKWT, BTKC481S, or BTKC481R mutations.22,52,53 It has been evaluated as a single agent in previously treated patients with WM with or without prior exposure to covalent BTK inhibitors as shown in Table 8.22,53 The ORR (88% vs 81%) were similar but the MRR (88% vs 67%) rates of nonexposed to covalent BTKi patients versus covalent BTK inhibitor pretreated patients were higher; however, pirtobrutinib monotherapy was able to overcome resistance to covalent BTKis. Importantly, the toxicity profile was quite favorable with very low rates of atrial arrhythmias or other covalent BTKis-related toxicities. Other ongoing clinical trials of pirtobrutinib either as monotherapy or in combination with other regimens in previously treated WM patients are shown in Table 9. Nemtabrutinib is another reversible, noncovalent BTK inhibitor which is investigated in B-cell malignancies.54,55 In WM has been evaluated as monotherapy and several trials are also ongoing as shown in Table 9.
Noncovalent BTK inhibitors in WM.

Referred to all pts with B-cell malignancies.
Most common (⩾5%).
BTK, Bruton tyrosine kinase; Gr, grade; mo, months; MRR, major response rate; n, number; NA, not available; No, number; ORR, overall response rate; PFS, progression-free survival; PR, partial response; pts, patients; RR, relapsed/refractory; cBTKi, covalent BTK inhibitors; TN, treatment naïve; VGPR, very good partial response; WM, Waldenström macroglobulinemia.
Other clinical trials of noncovalent BTK inhibitors in WM.

WM patients treated with a prior BTK inhibitor-containing regimen.
Experimental: Phase II (Pirtobrutinib monotherapy) cohort 5.
BTK, Bruton tyrosine kinase; RR, relapsed/refractory; WM, Waldenström macroglobulinemia.
BTK inhibitor combination therapies
Ibrutinib has been evaluated in combination with other regimens as shown in Table 1. In the iNNOVATE trial patients with symptomatic WM, either previously untreated or previously treated but nonresistant to rituximab therapy, were randomized 1:1 to ibrutinib–rituximab and placebo-rituximab. 56 The ibrutinib–rituximab cohort achieved substantially higher MRR and longer PFS compared to the rituximab-placebo cohort, with a 48-month PFS rate of 71% versus 25%. 57 The combination of ibrutinib and rituximab seemed to be “agnostic” of prior treatment (although all patients were sensitive to rituximab) and to genotype status, so that responses and PFS were similar for patients harboring CXCR4 mutations and even among those with MYD88 wild type in whom ibrutinib as a single agent has not favorable outcomes. 27 Although there were limitations in the sensitivity of the methods for detecting MYD88 and CXCR4 mutations, it is likely that addition of rituximab can overcome the detrimental effect of CXCR4 mutations when ibrutinib is used. Whether addition of rituximab to ibrutinib can improve the poor efficacy of ibrutinib monotherapy in MYD88WT is subject to further investigation.
Ibrutinib was also investigated in combination with the CXCR4-antagonist ulocuplumab in a small study of patients without previous exposure to BTK inhibitors. The combination led to a 2-year PFS of 90%, but despite the favorable results, ulocuplumab was discontinued by the manufacturer. 58
Venetoclax is the first-in-class BCL-2 inhibitor and fixed-duration combination with ibrutinib has shown very high efficacy with high rates of MRD negative and CRs in patients with CLL. 59 The fixed-duration combination of ibrutinib with venetoclax was evaluated in a phase II clinical trial that enrolled 45 previously untreated WM patients. 60 The primary endpoint was the attainment of VGPR; however, only 42% of patients achieved VGPR and no patient reached complete response (CR). The toxicity profile of the combination was problematic: four cases of ventricular arrhythmia occurred, two of them were fatal, leading to the termination of the study.
Based on in vitro data, combinations of ibrutinib with proteasome inhibitors could be a very active option, if proven safe and effective in clinical testing. 61 The combination of bortezomib–rituximab–ibrutinib has been explored by the European Consortium for Waldenstrom’s macroglobulinemia (ECWM) group (ECWM-2 study) and an ongoing study by the same group also compares ibrutinib monotherapy versus ibrutinib in combination with carfilzomib (CZAR study). 3 The results of these studies are awaited soon. Other ongoing clinical trials of ibrutinib with a variety of other regimens, such as rituximab and oral proteasome inhibitors, are shown in Table 2.
Zanubrutinib has also been investigated as combination therapy. Yu et al. 62 presented results from treatment-naïve patients who received the combination of zanubrutinib, ixazomib, and dexamethasone. Among 19 evaluable patients, all of them responded, while MRR and VGPR were attained in 94.7% and 42.1%, respectively. At a median follow-up of 12.3 months, all patients were alive; however, no CRs were reported.
Acalabrutinib is also investigated in a fixed-duration combination with bendamustine and rituximab (BR) as frontline therapy63,64; the regimen includes six cycles of BR and 12 months of acalabrutinib. An interim analysis that included the first 30 patients demonstrated a MRR and VGPR of 88% and 70%, respectively, after combination therapy. 64 At the end of the 6-month monotherapy at 12th cycle, eight out of nine patients had achieved VGPR (89%), while one reached CR (11%). Two patients after the end of the planned treatment were followed-up and both remained in VGPR. Other ongoing clinical trials of acalabrutinib in combination with other regimens are shown in Table 6.
BTK degraders and BCL-2 inhibitors
Beyond BTK inhibitors, BTK degraders are also under investigation. With this approach the BTK protein is removed by ubiquitination and proteosomal degradation. 65 BTK degraders that are evaluated in WM include NX-2127 (NCT04830137), NX-5948 (NCT05131022), and BGB-16673 (NCT05294731 and NCT05006716) with the respective clinical trials being ongoing.
Furthermore, venetoclax is a BCL-2 inhibitor approved for CLL/SLL and acute myeloid leukemia, playing an essential role in cellular apoptosis. A phase II clinical trial (NCT02677324) evaluated venetoclax in previously treated patients with WM, 16 of which were exposed to BTK inhibitors. 66 The ORR was 84% and the median PFS was 30 months. No deaths were recorded, suggesting that venetoclax can be a safe and effective treatment option for pretreated patients with BTK inhibitors. 66 Another BCL-2 inhibitor, sonrotoclax (BGB-11417) is a next-generation BCL-2 inhibitor more selective than venetoclax. 67 It is evaluated either as combination with zanubrutinib in treatment-naïve patients with WM or as monotherapy in previously treated patients, as sequencing treatment option after exposure to BTK inhibitors. 67 The clinical trial is ongoing and the results are expected.
BTKs inhibitors and Bing–Neel syndrome
Bing–Neel syndrome (BNS) is a rare complication of WM which is characterized by the infiltration of the central nervous system with malignant WM cells. 68 Even though BNS remains incurable, BTK inhibitors have shown efficacy. Ibrutinib enters the blood–brain barrier and as monotherapy has reported efficacy in the treatment of BNS.69,70 Castillo et al. in a multicenter study reported that patients receiving ibrutinib as monotherapy either in frontline or in relapsed settings, achieved symptomatic and radiologic improvement in 85% and 60%, respectively. The 2-year event-free survival (EFS) rate on ibrutinib was 80%, with the 5-year survival rate being 86%. Tirabrutinib has also been given to patients with BNS, revealing promising results either in frontline or as second or later line therapy.71–73 Zanubrutinib seemed effective in a female patient with BNS, however, further investigation is required. 74
Single agent or combinations of BTK inhibitors?
Current data indicate that covalent BTK inhibitors are the most effective single agents in WM, associated with very high rates of ORR and MRR, thus, being a favorable, chemotherapy-free treatment option, but still cannot achieve CRs and require continuous therapy. Combination regimens based on covalent BTKis should thus be explored to (A) limit the duration of therapy (i.e., fixed duration), (B) improve CR or VGPR rates, or (C) overcome resistance associated with specific genotypes. Most phase II and III studies have evaluated single-agent BTKis and only a few have explored combinations, most of which are still ongoing. In the iNNOVATE study the combination of ibrutinib with rituximab was compared to placebo–rituximab; however, there are no direct comparisons of ibrutinib plus rituximab versus ibrutinib monotherapy. Comparing the efficacy of ibrutinib monotherapy, from other studies in WM, to the experimental arm of iNNOVATE, the combination therapy does not seem to have substantially better efficacy. However, there may be patient groups which may benefit with combination over monotherapy with ibrutinib. Genomics can affect the choice of a combination regimen, since data from iNNOVATE indicate that addition of rituximab may be of benefit in patients with MYD88L265P/CXCR4mut genotypes. In the same study, the combination seemed to be agnostic of MYD88WT, but the numbers are limited. It is also notable that ibrutinib reduced the infusion-related reactions to rituximab and need for plasmapheresis by reducing the risk of IgM flare, associated with rituximab. Whether these observations seen with rituximab and ibrutinib may also be applicable to newer (and more active) covalent BTK inhibitors has not yet been explored. The combination of chemoimmunotherapy (such as BR) with covalent BTK inhibitors (acalabrutinib) is a chemotherapy-based, fixed-duration regimen and despite the high response rates (but in a small number of patients in the BRAWM study) cannot be proposed yet, since (A) there is limited data and (B) there was a lack of benefit of such approaches in CLL/SLL. 75 A major potential advantage of a combination of BTKi with immunochemotherapy could be the opportunity to offer a fixed duration of therapy. Since these studies are ongoing, such combination cannot be proposed outside their context. Their results, including additional assessment of the disease, for example, the use of assays to assess minimal residual disease, are eagerly awaited.
Combinations of BTKis with proteasome inhibitors could be an interesting option, which is explored in several trials (with either bortezomib, Carfilzomib, or ixazomib). The results of these studies are awaited, but adopting such regimens would require a substantial improvement in VGPR and CR rates over BTK monotherapy and convincing evidence that such combinations can overcome the detrimental effects of CXCR4 mutations. Treatment interruption of BTKi can lead to rebound phenomena; hence, patient’s desire and availability concerning the schedule of treatment is another factor that should be also taken into consideration. The nonchemotherapy containing combination of ibrutinib with venetoclax fell short of the expectations: the toxicity was significant, but there were also no CRs and the VGPR rates did not reach the primary end point target in the phase II study by Castillo et al. 60 Such combinations could be more successful with second-generation covalent BTK inhibitors or with noncovalent BTK inhibitors, but we still need well designed clinical trials to prove their efficacy and safety. Given that WM is a disease with an indolent course in most cases, with the majority of patients being elderly, often having several co-morbidities, any combination should prioritize safety. However, for patients with IgM-related complications for which elimination of IgM is critical, combination therapies could provide an option, if CR rates are significantly higher than with standard approaches. Our opinion is that current data support the use of BTKis as monotherapy (preferably of second-generation BTKIs such as zanubrutinib due to their better toxicity profile), until we have convincing data to support specific combinations.
Is there an optimal sequence of BTK inhibitors?
BTK inhibitors can be part of the first or subsequent line regimen and have been approved for both indications. In first-line therapy, the current choice is between continuous BTK inhibitor therapy or fixed-duration chemoimmunotherapy. Both choices have excellent outcomes and patients with WM are expected to live a long, and with good quality, life. However, in both pathways (continuous BTK inhibitor therapy or fixed-duration chemoimmunotherapy) relapses will occur or (with BTK inhibitors) intolerance may require treatment discontinuation. The characteristics of the patients and of the disease are critical for the choice of primary therapy. The presence of certain WM-related complications (such as hyperviscosity, neuropathy, cytopenias, AL amyloidosis, BNS), the presence and severity of co-morbidities (cardiovascular, renal, infectious, etc.), the genetics of the disease (i.e., presence or absence of MYD88L265P, and if available of CXCR4 and TP53 mutations), logistics, costs, and patients’ personal beliefs and preferences are critical factors for the choice of one pathway over the other. The role of inflammation is still under investigation in WM and may have significant implications. Studies have shown that patients with inflammatory markers such as C-reactive protein treated with chemoimmunotherapy tend to have shorter time to next treatment, poorer PFS, OS, and EFS compared to patients without chronic inflammation.76–79 Even though the data of the role of BTK inhibitors in these patients are not enough, Debureaux et al. concluded that BTKs are effective in patients with inflammatory WM. Hence, in this group of patients BTKs inhibitors could be superior to chemoimmunotherapy. 76
We have several lines of data which show that patients who have failed chemoimmunotherapy or even patients who are resistant to rituximab can be very effectively salvaged with BTK inhibitors. It is notable that the efficacy of BTKi, including ibrutinib and newer BKTis, seems to be similar in previously untreated and previously treated patients with WM, especially among non-rituximab refractory and nontransformed patients. Data regarding the outcomes of patients after failure of BTK inhibitors are less. There are retrospective studies which show that such patients can be effectively salvaged with the use of chemoimmunotherapy and proteasome inhibitors-based combinations, but we now have prospective data showing the activity of noncovalent BTK inhibitors, venetoclax, and of the upcoming BTK degraders. Thus, even patients failing BTK inhibitors there are treatment options available. Given that chemoimmunotherapy could provide a long treatment-free interval for many patients, who could then be salvaged very effectively, if needed by BTKi, we would advocate an approach in which the primary choice would be fixed-duration immunochemotherapy, for patients eligible for both. The toxicity profile of each approach should be a major factor to be considered, especially cardiovascular and hemorrhagic complications, logistic, and out-of-pocket patient costs. Another approach which has not been explored extensively is the use of sequential therapy with BTK inhibitors and chemoimmunotherapy; however, the indications for such an approach are extremely limited. Finally, a practical issue is the sequence of BTK inhibitors in patients who are intolerant to the initial BTKi regimen. This is a common situation in clinical practice and many patients switch from first to second-generation covalent BTK inhibitors if they are intolerant to the initial therapy. Today, with the availability of noncovalent BTK inhibitors we have an additional option for such patients. Given the available data for covalent BTK inhibitors our approach is to use zanubrutinib over ibrutinib due to a more favorable toxicity profile. We do not advocate for use of noncovalent BTK inhibitors before covalent ones, since there is limited data for pirtobrutinib in BTK-naïve patients. Pirtobrutinib is a reasonable salvage option for patients failing covalent BTK inhibitors, especially among those with documented C481 mutations. Targeting BCL-2 pathway in patients failing BTKi is also important. Although the data are still limited, the available results with venetoclax are promising, while more selective, next generation BCL-2 inhibitors such as sonrotoclax are investigated. 67 Both venetoclax and pirtobrutinb seem reasonable options for patients failing covalent BTKis; whether a class switch (i.e., from BTK inhibition to BCL-2 inhibition) may be associated with better results is unknown, and the choice is based on the toxicity profile and availability of these drugs. The introduction of BTK degraders will further increase options while combinations (e.g., BCL-2 inhibitors with BTK degraders) may offer a fixed-duration chemotherapy-free regimen.
Conclusion
BTK inhibitors are an essential therapy for the management of WM. Until more data are available, covalent BTKis should be used as continuous therapy without interruptions, unless clearly indicated. Use of fixed-duration chemoimmunotherapy is our preferred first-line approach for most patients. For patients starting on a BTK inhibitor, we prefer to use approved second-generation cBTKis due to their toxicity profile, as single agents. Sequential use of different covalent BTKis can be considered if there is intolerance, but not recommended for patients with BTKi refractory disease. Noncovalent BTKi and BCL-2 inhibitors provide a new option to sequence covalent BTKis in cases of intolerance and of resistance. BTKis are the treatment to consider for special populations, such as those with BNS, rituximab resistance, bulky disease, and those in need for rapid IgM reduction to manage complications such as hyperviscosity syndrome (along with plasmapheresis).