
Abstract
To report the management and outcome of an elderly, high-risk lymphoplasmacytic lymphoma/Waldenström macroglobulinemia patient presenting with severe symptomatic anemia, marked hyperleukocytosis, a complex karyotype, dual MYD88/CXCR4 mutations, and significant comorbidities, highlighting the therapeutic challenges and the efficacy of targeted therapy after chemoimmunotherapy intolerance. This case report details a 78-year-old male diagnosed with high-risk lymphoplasmacytic lymphoma/Waldenström macroglobulinemia (MYD88 L265P+, pathogenic CXCR4+, complex karyotype: 46,X,-Y,add(6)(q224),+18/47,XY,+18/47,X,Y,+4,+18/48,XY,+4,+18/48,XY,+3,+18/46,XY) and significant comorbidities including hypertension and suspected cardiac amyloidosis. Initial treatment with bendamustine–rituximab was administered but led to significant toxicity. Subsequently, therapy was switched to the second-generation Bruton’s tyrosine kinase inhibitor, zanubrutinib. Clinical progress, hematologic response, and adverse events were monitored. Initial treatment with bendamustine–rituximab was discontinued due to severe toxicity, including a grade 3 infusion reaction and subsequent neutropenic fever. Following the switch to zanubrutinib, he achieved a sustained partial hematologic response and clinical improvement. Disease-related complications (hyperviscosity retinopathy) and treatment-related adverse events (neutropenia) occurred but were managed appropriately while continuing zanubrutinib. Zanubrutinib demonstrated efficacy and manageable toxicity in an elderly, high-risk lymphoplasmacytic lymphoma/Waldenström macroglobulinemia patient with a complex karyotype, dual MYD88/CXCR4 mutations, hyperleukocytosis, and significant comorbidities, following intolerance to standard chemoimmunotherapy. This case supports the use of targeted Bruton’s tyrosine kinase inhibition in achieving favorable outcomes in complex lymphoplasmacytic lymphoma/Waldenström macroglobulinemia presentations and contributes to understanding the significance of complex karyotypes in the era of novel therapies.
Introduction
Lymphoplasmacytic lymphoma (LPL) is a malignancy of mature B-cells characterized by infiltration of the bone marrow by small lymphocytes, plasmacytoid lymphocytes, and plasma cells. 1 When associated with a monoclonal IgM paraprotein, it is termed Waldenström macroglobulinemia (WM). 2 WM typically affects older individuals, with symptoms arising from tumor infiltration (cytopenias, organomegaly) or the IgM paraprotein (hyperviscosity, neuropathy, cryoglobulinemia). 3 While typically presenting as an indolent disease, a subset of patients can exhibit aggressive features. Marked hyperleukocytosis, as seen in this case, is an uncommon finding in WM and often signals a higher tumor burden or more aggressive disease biology. The MYD88 L265P mutation is a key diagnostic marker found in over 90% of WM cases, 4 while CXCR4 mutations occur in approximately 30%–40% of patients and can influence disease behavior and treatment response. 5 Therapeutic strategies range from watchful waiting for asymptomatic patients to chemoimmunotherapy or targeted therapies, such as Bruton’s tyrosine kinase (BTK) inhibitors, for symptomatic disease. 6 Managing elderly patients often requires balancing efficacy with tolerability due to comorbidities and potential treatment toxicities. Complex karyotypes (CKs), defined by multiple chromosomal abnormalities, 7 are generally associated with adverse outcomes in hematological malignancies, 8 but their specific impact in WM, especially in the era of targeted therapies, is less defined. The co-occurrence of a CK with dual MYD88/CXCR4 mutations presents a unique therapeutic challenge, as this triad of high-risk features is not well characterized in the literature. Here, we present a case of high-risk LPL/WM with unusual features, including hyperleukocytosis and a CK, successfully managed with zanubrutinib, and review the literature regarding the prognostic significance of CK in WM.
Case presentation
A late 70s retired male from Shanghai, China, with a 20-year history of grade 3 (very high risk) hypertension (well-controlled on nifedipine and irbesartan), presented with a 9-month history of progressively worsening dizziness, fatigue, and gait instability. Initial evaluation at an external hospital revealed severe anemia (hemoglobin (HGB) 64 g/L) and marked leukocytosis (white blood cell count (WBC) 58.0 × 109/L) with 90.9% lymphocytes, predominantly mature small lymphocytes on peripheral blood smear. Abdominal ultrasound showed splenomegaly, fatty liver, cholelithiasis, and renal cysts, with no significant lymphadenopathy. Chest computed tomography (CT) showed minor pulmonary lesions and a right axillary lipoma. Brain magnetic resonance imaging indicated age-related changes and chronic lacunar infarcts. Further investigations at a tertiary referral center approximately 1 week later confirmed severe anemia (HGB 58 g/L), persistent hyperleukocytosis (WBC 56.2 × 109/L, 86.1% lymphocytes), elevated total protein (101 g/L), low albumin (3.3 g/dL), and a decreased albumin/globulin ratio (0.49), suggesting paraproteinemia. Serum protein electrophoresis and immunofixation identified a monoclonal IgM kappa paraprotein, with a baseline serum IgM level of 90.9 g/L, confirming the diagnosis of WM and indicating a very high disease burden. Peripheral blood flow cytometry identified a large (79.3%) population of abnormal mature B-lymphocytes with the immunophenotype: CD19+, CD5−, CD10(low+), CD23−, CD22(dim+), CD79b+, CD20(strong+), FMC7+, CD200+, CD43(partial+), Kappa light chain restricted, Lambda−, CD103−, CD11c−, CD25+, CD38−, CD138−. This phenotype was suggestive of a B-cell non-Hodgkin lymphoma, potentially LPL/WM or marginal zone lymphoma (MZL). The flow cytometry analysis demonstrating the gating strategy and aberrant B-cell immunophenotype is shown in Figure 1.

Flow cytometry analysis of peripheral blood.
A positron emission tomography (PET)-CT scan performed shortly thereafter demonstrated widespread disease (Figure 2), including multiple enlarged lymph nodes with mild-to-moderate Fludeoxyglucose (FDG) uptake (max standardized uptake value (SUV) 5.3 in left renal hilum), splenomegaly (15 cm) with heterogeneous uptake (max SUV 4.4) and infarcts, hepatomegaly, and diffuse bone marrow hypermetabolism (max SUV 3.4). Other findings included a metabolically active pulmonary ground-glass opacity (SUVmax 3.1) in the left upper lobe, suspicious for lymphomatous involvement, pituitary uptake, sinusitis, prostatic hyperplasia, and spinal degenerative changes.

PET-CT scan at baseline. Representative images show mild-to-moderate FDG uptake in the left renal hilum and the left lung.
Due to worsening fatigue and inability to walk, the patient was admitted to a specialized medical center approximately 3 weeks after the investigations at the referral center. Initial management included supportive care with blood transfusions, hydration, and dexamethasone. Bone marrow aspirate and biopsy performed shortly after admission showed a markedly hypercellular marrow (95%) extensively infiltrated (85%–91%) by small lymphocytes, suppressing normal hematopoiesis (Figure 3).

Bone marrow aspirate morphology. The marrow is markedly hypercellular and diffusely infiltrated by a monotonous population of small lymphocytes with condensed chromatin, scant cytoplasm, and occasional plasmacytoid features, consistent with lymphoplasmacytic lymphoma.
Flow cytometry on the bone marrow aspirate confirmed the abnormal B-cell population (79.0%), which was immunophenotypically identical to the population identified in the peripheral blood. The initial interpretation of the bone marrow biopsy was consistent with an indolent B-cell lymphoma/leukemia involving the marrow. A concurrent fine-needle aspiration biopsy of a right inguinal lymph node initially suggested MZL, pending molecular studies. During this admission, the patient developed acute urinary retention secondary to benign prostatic hyperplasia (BPH), requiring an indwelling catheter, and elevated B-type natriuretic peptide (BNP) levels treated with diuretics. Concurrently, he exhibited signs of cardiac strain with significantly elevated N-terminal pro-BNP levels peaking at 5756 pg/mL (reference range <300 pg/mL), while high-sensitivity troponin levels remained within the normal range. These findings, in conjunction with echocardiographic evidence of myocardial thickening, led to a clinical diagnosis of suspected cardiac amyloidosis. He was treated with diuretics to manage fluid overload.
Based on a likely diagnosis of a B-cell malignancy requiring treatment (severe symptomatic anemia, high tumor burden including hyperleukocytosis), chemoimmunotherapy with the bendamustine–rituximab (BR) regimen was initiated approximately 2 weeks after admission (rituximab 400 mg d1, 293 mg d2; bendamustine 90 mg d3, d4). The treatment was complicated by a severe rituximab infusion reaction (grade 3), manifesting as acute-onset tachycardia, atrial fibrillation, and diaphoresis, requiring interruption and intensive management with steroids, antihistamines, and digoxin. Approximately 4 days after starting the regimen, the patient developed fever (up to 39.2°C), cough, and sputum production, treated empirically for infection with broad-spectrum antibiotics (meropenem, levofloxacin) and nebulization. Given the severity of both the infusion reaction and the subsequent myelosuppression in this frail patient, the BR regimen was deemed intolerable. He also experienced minor rectal bleeding (attributed to hemorrhoids) and recurrent urinary retention managed with an indwelling catheter and doxazosin.
Pathology review and molecular results became available approximately 3 weeks after starting BR. Both bone marrow and lymph node biopsies were reviewed by pathology experts at the referral center and definitively diagnosed as LPL. Crucially, molecular analysis detected monoclonal IGH gene rearrangement, the MYD88 L265P mutation, and a pathogenic CXCR4 mutation. Further genetic testing revealed a complex karyotype: 46,X,Y,add(6)(q224),+18/47,XY,+18/47,X,Y,+4,+18/48,XY,+4,+18/48,XY,+3,+18/46,XY. This indicates multiple clonal chromosomal abnormalities, including loss of Y chromosome, addition on chromosome 6q, trisomy 18, trisomy 4, and trisomy 3 in various clones (Figure 4).

Representative karyograms from the patient demonstrating components of the complex karyotype.
Fluorescence in situ hybridization (FISH) was negative for D13S319 (13q deletion), RB1, ATM, chromosome 12 polysomy, and TP53 (p53 deletion). The International Prognostic Scoring System for WM (IPSSWM) score was calculated as 4, indicating High Risk, based on the following parameters: Age >65 years (1 point), hemoglobin ⩽11.5 g/dL (1 point), β2-microglobulin >3 mg/L (patient value: 7.74 mg/L; 1 point), and serum monoclonal IgM >70 g/L (patient value: 90.9 g/L; 1 point). The platelet count was 205 × 10⁹/L (>100 × 10⁹/L, 0 points). The revised IPSSWM (rIPSSWM) score was also 4 (incorporating LDH >ULN (implied) and albumin <3.5 g/dL (patient’s albumin 3.3 g/L)), classifying him into the Very High-Risk group. The diagnosis of LPL/WM was firmly established.
Given the patient’s intolerance to the BR regimen and his molecular profile, the treatment strategy was changed approximately 4 weeks after starting BR to the BTK inhibitor zanubrutinib 160 mg twice daily. Following the initiation of zanubrutinib, the patient’s condition gradually improved. However, approximately 11 days after starting zanubrutinib, he reported decreased vision. Ophthalmology consultation revealed bilateral retinal hemorrhages, macular edema, and shallow choroidal detachment, consistent with hyperviscosity retinopathy secondary to WM. As systemic therapy with zanubrutinib was already underway to address the underlying cause of paraproteinemia, and the patient’s visual symptoms were not acutely progressing to blindness, a decision was made to continue targeted therapy and monitor closely rather than proceed with immediate plasmapheresis. Zanubrutinib was continued as the primary treatment for the underlying WM. The patient continued zanubrutinib therapy during subsequent follow-ups over the next 11–12 months.
The patient experienced significant toxicity with the initial BR regimen. Following the switch to zanubrutinib, he achieved a sustained partial hematologic response and clinical improvement. Disease-related complications (hyperviscosity retinopathy) and treatment-related adverse events (neutropenia) occurred but were managed appropriately while continuing zanubrutinib. His M-protein level showed a gradual decline from 7.5 g/L (measured ~6 months post-zanubrutinib) to 6.91 g/L (~9 months) and further to 6.68 g/L (~10 months), indicating a partial hematologic response. Changes in key clinical parameters over time are listed in Table 1.
Changes in key clinical parameters over time.
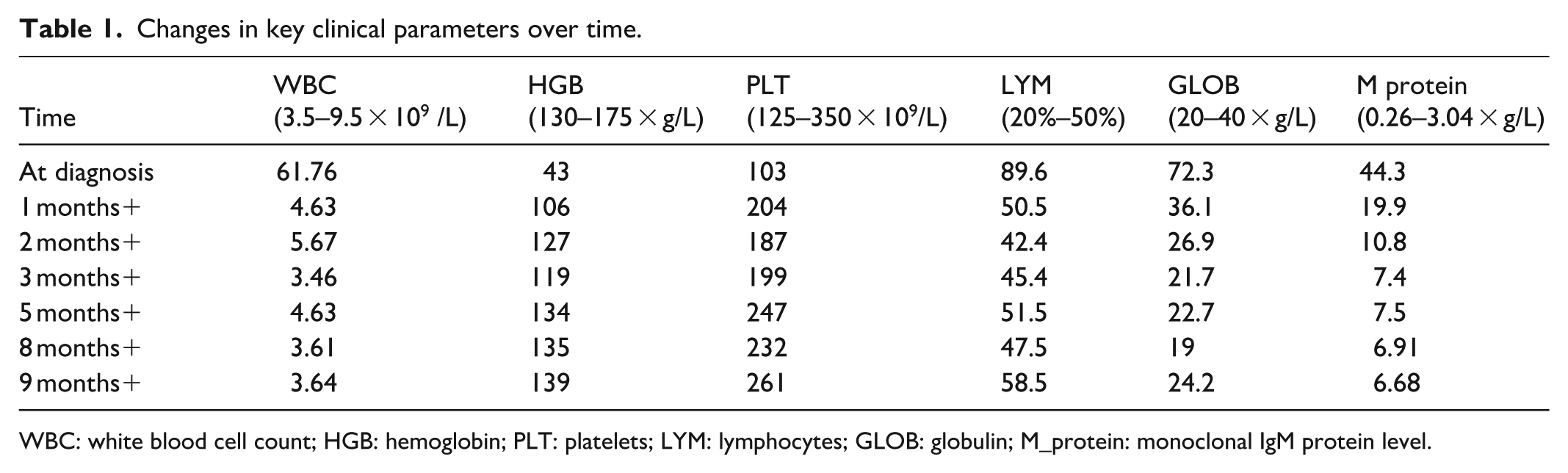
WBC: white blood cell count; HGB: hemoglobin; PLT: platelets; LYM: lymphocytes; GLOB: globulin; M_protein: monoclonal IgM protein level.
His overall clinical condition stabilized; admission notes from approximately 6 months post-zanubrutinib indicated fair energy, appetite, and sleep. Physical examination at that time revealed no definitively palpable lymphadenopathy or hepatosplenomegaly, potentially reflecting treatment response reducing organ size below palpable limits. Neutropenia occurred during treatment, necessitating supportive care with granulocyte colony-stimulating factor (G-CSF). An echocardiogram finding (strong echo on a coronary valve leaflet) prompted cardiology consultation, recommending cardiothoracic surgery evaluation and close follow-up due to suspicion of cardiac involvement (amyloidosis was listed in the final diagnoses). As of the last documented visit, approximately 11.5 months after initiating zanubrutinib (a brief admission for medication continuation), the patient was reported to be in stable condition (“general condition fair”) and was discharged the same day, continuing zanubrutinib 160 mg BID along with prophylactic medications (trimethoprim–sulfamethoxazole, acyclovir) and supportive care (including Leucogen, a derivative of cysteine, which can promote the growth and maturation of granulocytes in the bone marrow and stimulate the proliferation of leukocytes and platelets). He remains under close monitoring for disease status, treatment toxicity (cytopenias), and management of comorbidities (cardiac condition, BPH, hypertension). His final diagnosis list included: LPL/WM (high/very high risk, MYD88+/CXCR4+), suspected cardiac amyloidosis, BPH, hypertension, pulmonary lesions, splenomegaly.
Discussion and literature review
This case illustrates several critical aspects of managing elderly patients with high-risk LPL/WM, particularly those with complex presentations including hyperleukocytosis, CK, and significant comorbidities. The presentation with severe anemia and fatigue is typical for symptomatic WM. 9 The diagnostic workup highlights the importance of integrating morphology, extensive immunophenotyping (flow cytometry), imaging (PET-CT), molecular analysis (MYD88/CXCR4 mutations), and cytogenetics. The initial differential diagnosis including MZL underscores the challenges in distinguishing indolent B-cell lymphomas without complete data. 10 Confirmation of the MYD88 L265P mutation was essential for the definitive diagnosis of LPL/WM. 11 The marked hyperleukocytosis is an atypical feature, potentially reflecting a more aggressive “leukemic phase” of the disease. This presentation could be driven by underlying biology, such as the pathogenic CXCR4 mutation, which is known to influence cell trafficking and may promote the egress of malignant cells from the bone marrow into the peripheral blood. The coexistence of a pathogenic CXCR4 mutation places the patient in a higher-risk category. 12
The choice of zanubrutinib as second-line therapy was multifactorial. Following intolerance to BR, a BTK inhibitor was a rational choice given the patient’s MYD88 L265P mutation. Among BTK inhibitors, zanubrutinib was specifically selected over the first-generation inhibitor ibrutinib due to its superior selectivity and more favorable safety profile. Ibrutinib is associated with higher rates of off-target toxicities, including atrial fibrillation and bleeding. Given our patient’s history of treatment-induced atrial fibrillation, preexisting severe hypertension, and suspected cardiac amyloidosis, zanubrutinib presented a safer therapeutic option with a lower risk of exacerbating his cardiovascular comorbidities. 6
A particularly noteworthy aspect of this case is the presence of a CK. CK, generally defined by the presence of multiple chromosomal abnormalities, 13 is often associated with poor prognosis and aggressive disease features in various hematologic malignancies, including chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), MDS, and mantle cell lymphoma (MCL). 8 However, a universally accepted definition remains elusive. Definitions range from two or more abnormalities in myeloma to three or more in acute myeloid leukemia (AML), MDS, and often CLL, 14 with some classifications further distinguishing low, 3 intermediate, 4 and high-complex (⩾5) karyotypes. 7 In WM specifically, CK has been defined as ⩾3 clonal aberrations detected by chromosome banding analysis, 15 with high CK defined as ⩾5 aberrations. 16 Our patient, with multiple clones exhibiting combinations of −Y, add(6q), +3, +4, and +18, clearly meets the definition of CK and potentially high CK depending on the clone analyzed.
Prior to the widespread use of novel therapies, investigations suggested CK, particularly high CK (⩾5 aberrations), was likely an adverse prognostic factor in WM. 15 An abnormal karyotype was correlated with inferior overall survival (OS) and progression-free survival (PFS) in some studies. 17 Specific abnormalities common in WM, such as del(6q) 18 and del(17p), were also linked to poorer outcomes. 19
The treatment landscape for WM has been transformed by novel agents like BTK inhibitors (ibrutinib, zanubrutinib, acalabrutinib), proteasome inhibitors (bortezomib), and BCL-2 antagonists (venetoclax). 8 The question arises whether the prognostic impact of CK persists in this new era. Studies have yielded somewhat mixed results. A retrospective analysis by Krzisch et al. found high CK (⩾5 aberrations) remained associated with shorter PFS. 15 Another study also found abnormal karyotypes (including CK) correlated with inferior OS and PFS, particularly in older patients, even within an era incorporating novel therapies. 17 An analysis by Blanco et al. suggested chromosomal abnormalities, including complex ones (defined as ⩾2), were adverse predictors. 20 Table 2 summarizes findings on the prognostic impact across eras.
Prognostic impact of complex karyotype in WM across different treatment eras.
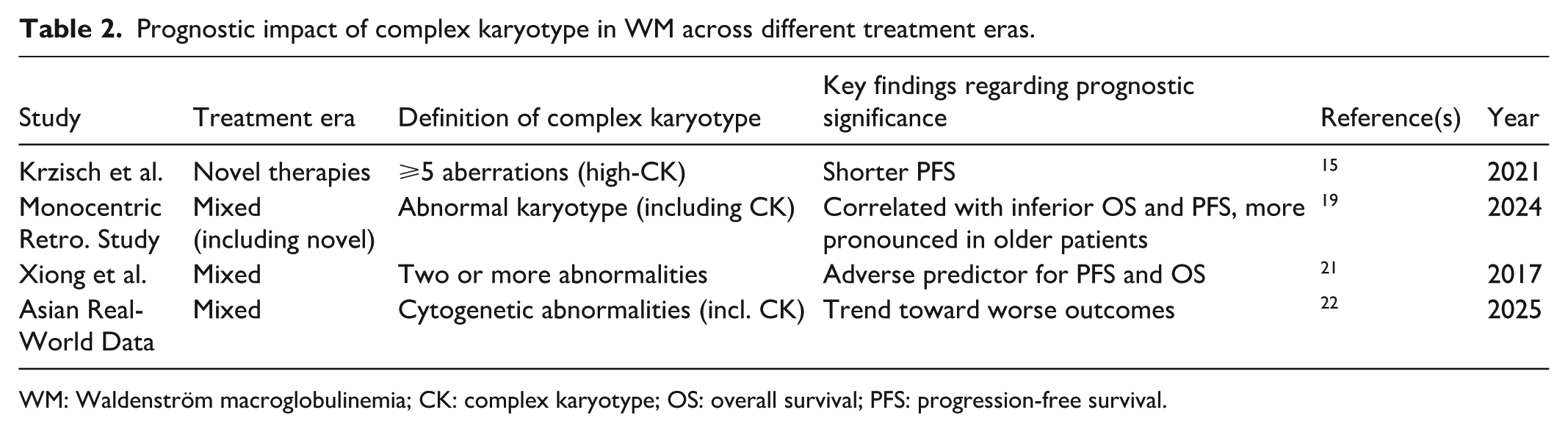
WM: Waldenström macroglobulinemia; CK: complex karyotype; OS: overall survival; PFS: progression-free survival.
However, expert commentary suggests that while CK predicts lower efficacy with chemoimmunotherapy, its negative impact might be attenuated with BTK inhibitors and BCL-2 inhibitors, although earlier relapses may still occur. 23 This aligns with findings in CLL, where the adverse impact of CK seen with chemoimmunotherapy may be lessened, though not entirely eliminated, by novel agents. 24
Within a CK, specific abnormalities may retain prognostic weight. Del(17p)/TP53 abnormalities remain a strong adverse marker in related B-cell cancers treated with novel agents and are associated with shorter PFS in WM. 25 Del(6q) has also been consistently linked to adverse prognosis and shorter survival in WM. 18 The prognostic significance of specific abnormalities is summarized in Table 3.
Prognostic significance of specific chromosomal abnormalities within complex karyotype in WM and novel therapies.

WM: Waldenström macroglobulinemia; BTKi: Bruton’s tyrosine kinase inhibitor; CK: complex karyotype; CLL: chronic lymphocytic leukemia; PFS: progression-free survival.
Our patient presents a compelling scenario: high-risk disease (IPSSWM/rIPSSWM 4) with a CK involving +3, +4, +18, −Y, and add(6q) (which may harbor similar prognostic implications to del(6q), 40), yet demonstrating a sustained partial response and clinical stability on Zanubrutinib. A timeline summarizing the patient’s complex clinical course is presented in Figure 5. This outcome occurred despite the add(6q) abnormality and high-risk clinical scores. The presence of a pathogenic CXCR4 mutation is known to be associated with resistance to ibrutinib and can lead to slower or less profound responses to BTK inhibition. 5 The clinical course of our patient—a gradual but sustained partial response rather than a rapid, deep remission—may indeed reflect the attenuating influence of his CXCR4 mutation on zanubrutinib’s efficacy. Crucially, FISH analysis was negative for high-risk deletions involving TP53 (17p), ATM (11q), and RB1/D13S319 (13q). This absence of specific high-risk FISH lesions, despite the numerical and structural complexity observed via conventional karyotyping, might be a key factor contributing to the favorable response to BTK inhibition in this case. It suggests that the quality of aberrations within a CK, not just the quantity, significantly influences prognosis, particularly with targeted therapy. The efficacy of zanubrutinib, a second-generation BTK inhibitor with a favorable tolerability profile, was evident after the patient experienced significant toxicity with BR, a common issue in older, comorbid patients.

Timeline of key clinical and therapeutic events.
This case also underscores the multifaceted nature of WM complications and management challenges. Hyperviscosity retinopathy requires ophthalmologic monitoring alongside systemic therapy. Treatment-related neutropenia necessitated G-CSF support. Furthermore, the concurrent management of significant comorbidities—severe hypertension, BPH causing urinary retention, and suspected cardiac amyloidosis—added significant complexity. The cardiac findings warrant ongoing investigation, as cardiac involvement can significantly impact prognosis.
Conclusion
This case report details the successful management of an elderly patient with high-risk, MYD88/CXCR4-mutated LPL/WM, presenting with hyperleukocytosis and harboring a CK, using zanubrutinib following intolerance to initial chemoimmunotherapy. The literature suggests that while CK, especially high CK or those containing specific high-risk lesions like del(17p) or del(6q), often indicates a poorer prognosis, the impact might be attenuated by novel agents like BTK inhibitors. Our case, demonstrating a sustained partial response despite a CK lacking high-risk FISH abnormalities, supports the notion that the specific composition of the CK is critical and that BTK inhibition can be effective in such patients. This highlights the importance of comprehensive diagnostics, including conventional cytogenetics and FISH/molecular testing, and demonstrates the effectiveness and relative tolerability of novel targeted agents like zanubrutinib in the complex management of elderly WM patients, even those with traditionally high-risk genetics including CK. Continued monitoring for disease progression, treatment toxicity, and comorbidity management remains essential. If the patient experiences disease progression on zanubrutinib, future therapeutic options could include BCL-2 inhibitor-based regimens, such as with venetoclax, or enrollment in clinical trials investigating novel therapeutic agents.
Footnotes
Ethical considerations
This work has been reported in line with the Case Report (CARE) criteria. Our institution has waived the requirement of ethical approval for reporting individual cases or case series.
Author contributions
JC, JZ, and CX contributed equally to this work. Conceptualization: JC, JZ, CX, JH, RL. Data curation: JC, JZ, CX, BZ. Formal analysis: JC, JZ, CX. Investigation: JC, JZ, CX, BZ. Methodology: JC, JZ, CX, JH, RL. Writing – original draft: JC, JZ, CX. Writing – review & editing: JC, JZ, CX, BZ, JH, RL. Supervision: JH, RL. Project administration: JH, RL. All authors read and approved the final manuscript and agree to be accountable for all aspects of the work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (82300235), the Shanghai Municipal Health Commission Talent Plan Youth Project (2022YQ031), and the Sanhang Talent program of Naval Medical University (25TPSL0003).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data and materials used in this study are available upon request from the corresponding author. No datasets were generated or analyzed during the current study.