
Abstract
Despite major therapeutic advancements in recent years, multiple myeloma (MM) remains an incurable disease with nearly all patients experiencing relapsed and refractory disease over the course of treatment. Extending the duration and durability of clinical responses will necessitate the development of therapeutics with novel targets that are capable of robustly and specifically eliminating myeloma cells. B-cell maturation antigen (BCMA) is a membrane-bound protein expressed predominantly on malignant plasma cells and has recently been the target of several novel therapeutics to treat MM patients. This review will focus on recently approved and currently in development agents that target this protein, including bispecific antibodies, antibody-drug conjugates, and chimeric antigen receptor T-cell therapies. In addition, this protein also serves as a novel serum biomarker to predict outcomes and monitor disease status for MM patients; the studies demonstrating this use of BCMA will be discussed in detail.
Keywords
Introduction
Multiple myeloma (MM) is the second most common hematological malignancy and accounts for roughly 2% of all cancer cases and related deaths within the United States. 1
MM is a bone marrow (BM)-based cancer that accounts for approximately 10% of all blood-based malignancies and is characterized by the production of monoclonal antibodies by malignant plasma cells (PCs).2–4 Commonly used drug classes for the treatment of MM include proteasome inhibitors (PIs), immunomodulatory agents (IMiDs), and monoclonal antibodies. 5 Despite the current large number of treatment options, MM patients are unable to maintain control of their disease indefinitely; and thus, they will require additional new therapeutic approaches for the long-term management of their disease.6,7 Moreover, there is a need for more effective ways to monitor these patients, especially for the 1%–5% of MM patients that lack the conventional markers, serum M protein (sMP) or serum-free light chains (sFLCs), to follow their disease progression.8,9 B-cell maturation antigen (BCMA), also known as CD269, is an ideal therapeutic target and serum biomarker for MM patients since it is expressed predominantly on mature B lymphocytes, especially plasma cells, and is shed into the blood with levels that are easily measurable 10 . Its overexpression and activation have been associated with MM in preclinical and clinical studies. 10
BCMA is a cell surface antigen and a member of the tumor necrosis factor receptor (TNFR) family. It exhibits a restricted expression pattern with immunolabeling studies indicating that it is predominantly expressed on PCs and is absent on other normal human tissues and human CD34(+) hematopoietic multipotential stem cells. 11 Within the long-lived BM PCs, BCMA is essential for survival; however, the same does not hold true for short-lived PCs or B-cells.12–14 Ligands for BCMA include a B-cell activating factor (BAFF) as well as a proliferation-inducing ligand (APRIL). The interaction of BCMA with its ligands is thought to play a role in both the survival and proliferation of MM cells as well as the immunodeficiency often observed among MM patients.15,16 In vitro and in vivo studies have shown that the overexpression of BCMA on MM cells leads to the aberrant activation of the BCMA/APRIL pathway and subsequent increase in tumor burden.15,17 Additionally, soluble BCMA (sBCMA) is produced through shedding of the protein from the cell surface and mediated by gamma secretase. 18 In vivo and in vitro experiments investigating sBCMA and BAFF showed that sBCMA binds to BAFF preventing it from engaging membrane-bound BCMA on normal PCs. This leads to a reduction in the differentiation of normal PCs, subsequently causing a decline in polyclonal antibody levels, thereby contributing to immunodeficiency. 16 Due to the specificity of elevated BCMA expression to malignant plasma cells and its presence in blood, BCMA has become a key target for development of new agents for MM as well as becoming a potential new biomarker for determining the prognosis and monitoring these patients.
The biological basis for targeting BCMA in MM is based on preclinical models showing murine xenografts that had BCMA overexpression grew faster than BCMA-negative controls. The overexpression of BCMA leads to amplification of genes critical for metastasis, osteoclast activation, immunosuppression, and growth. This receptor is also expressed on plasmacytoid dendritic cells which help aid in survival of MM cells in the BM.15–17
This review will provide an overview of the three currently approved therapeutic approaches targeting BCMA for the treatment of MM, including bispecific antibodies (BsAbs), antibody-drug conjugates (ADCs), and chimeric antigen receptor (CAR) T-cell therapies. This review will also examine the role of sBCMA as a prognostic and monitoring biomarker for MM patients.
Material and methods
A PubMed search was used to identify published data on BCMA starting from the dates of January 1, 2012, to October 1, 2023. PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines were used to comprehensibly report our review process (Figure 1; Supplemental Material). 19 Abstracts from major oncology and hematology conferences (such as the American Society of Hematology and the American Society of Clinical Oncology) within the same time frame were additionally used. BCMA-targeted therapies including BsAbs, ADCs, and CAR T-cell therapies are discussed in this study as well as studies that provide evidence for the use of sBCMA as a novel biomarker, both as a prognostic and monitoring tool. Terms used to find data were “BCMA,” “BCMA therapies,” “BCMA as a biomarker,” BCMA as a diagnostic marker,” “BCMA as a prognostic factor,” “CD269,” and “TNFRSF17” for therapeutic target and “multiple myeloma” for disease state.
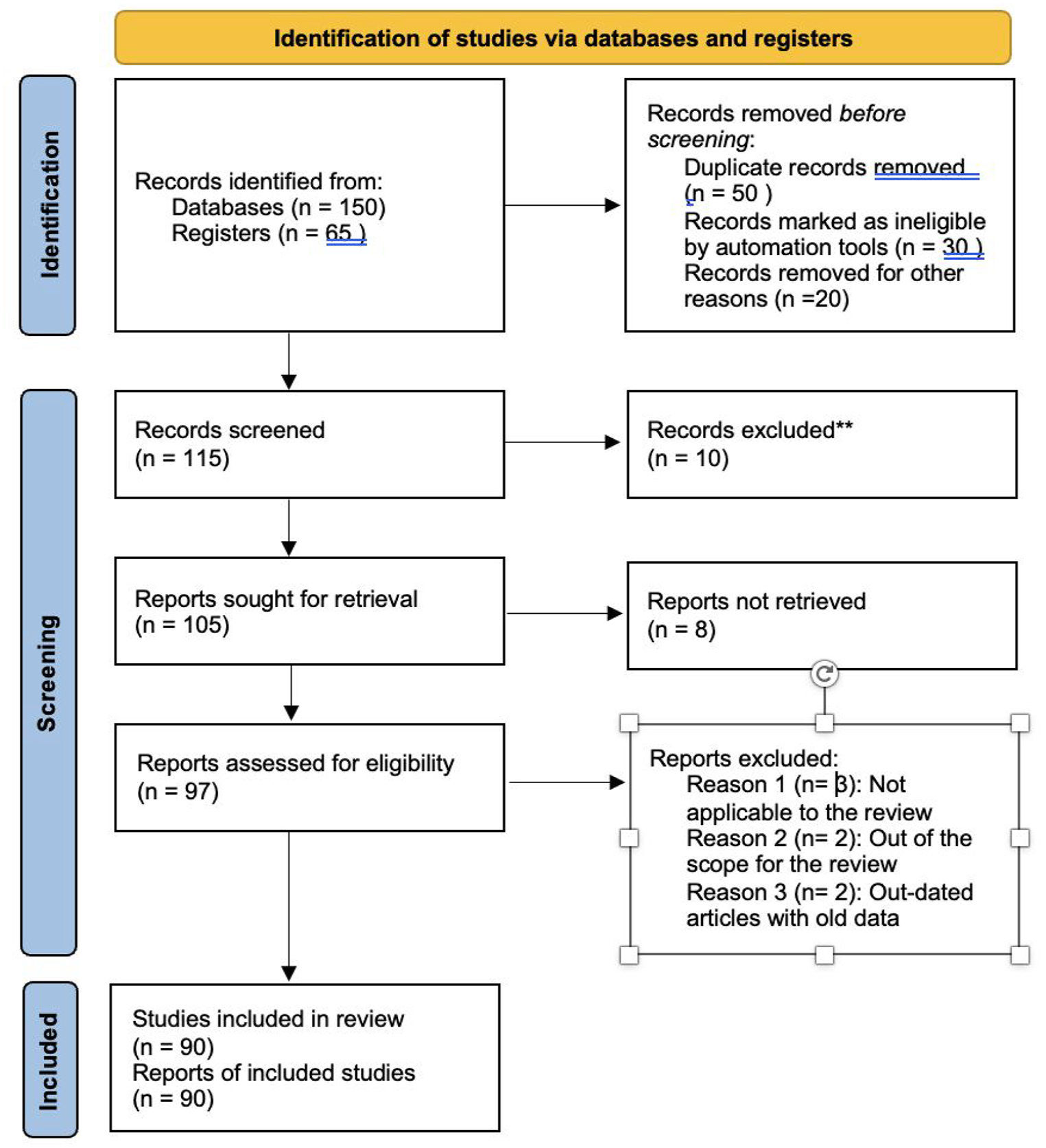
PRISMA flowchart.
Review of BCMA-related studies in MM
Bispecific antibodies
Current BsAbs for the treatment of MM are antibodies that have been engineered to bind two different epitopes, one on MM cells (BCMA) and the other on T-cells (CD3), in order to facilitate cell-to-cell interactions and induce cytolytic activity, a phenomenon known as a cytolytic immune synapse20,21 (Figure 2). There are currently two FDA-approved BCMA × CD3 BsAbs, teclistamab and elranatamab, with several other BsAb constructs currently being investigated for the treatment of relapsed/refractory multiple myeloma (RRMM) patients. BCMA × CD3 BsAbs in currently ongoing clinical trials include: (1) ABBV-383, (2) linvoseltamab, (3) alnuctanab, and (4) AMG420.

Schematic illustration of BCMA-targeted immunotherapies.
Teclistamab
Teclistamab (JNJ-64007957) is a humanized BCMA × CD3 BsAb produced by Janssen. A phase I/II MajesTEC-1 trial 22 involved 165 RRMM patients, 77.6% of which had triple-class refractory disease. Patients received a weekly subcutaneous injection of teclistamab (at a dose of 1.5 mg/kg of body weight) after receiving two step-up doses of 0.06 and 0.3 mg/kg during the first week of treatment. The overall response rate (ORR) was 63% with a median follow-up of 14.1 months. Measurable residual disease (MRD) negativity was achieved in 26.7% of patients and the median duration of response (DOR) was 18.4 months with an 11.4-month median progression-free survival (PFS). The most common adverse events (AEs) found in this study were cytokine release syndrome (CRS; 72.1%), neutropenia (70.9%), anemia (52.1%), and thrombocytopenia (40.0%). There was a high rate of infection observed (76.4%; grade 3 or 4, 44.8%) and neurotoxic events occurred in 24 patients, 5 of which were immune effector cell-associated neurotoxicity syndrome (ICANS). Despite the frequency and severity of AEs, the high rates of deep and durable responses achieved in this study led to the FDA approval of teclistamab in 2022. 22
Teclistamab has also been investigated when combined with talquetamab, a G protein-coupled receptor, class C, group 5, member D (GPRC5D)-targeted BsAb, in a phase Ib trial (RedirecTT-1 study). 23 The study enrolled 63 MM patients with RR disease who previously received a PI, IMiD, and anti-CD38 antibody therapy. The median age of patients was 67 years, and the median prior lines of therapy were 5 (1–11). Within this patient population, 33% had high-risk cytogenetics, 78% were triple-class refractory, 63% were penta-drug exposed, and 43% had extramedullary disease (EMD). The most common AEs were CRS (81%; 3% grade 3), neutropenia (76%; 75% grade 3/4), and anemia (60%; 43% grade 3/4). The ORR was 84% and 73% among patients with EMD. The rate of complete response (CR) or better than CR was 34% and 31%, respectively and median DOR has not yet been reached. 23
Elranatamab
Elranatamab (PF-06863135) is a humanized IgG2a BsAb targeting BCMA and CD3 and is produced by Pfizer (New York, New York). Promising results from the ongoing phase II MagnetisMM-3 trial (NCT04649359) have resulted in the recent FDA approval of this BSAb for RRMM patients.
24
In this trial, 123 RRMM patients received 76 mg elranatamab subcutaneously once weekly (QW) resulting in an ORR of 61% (75/123) and
ABBV-383
ABBV-383 is a BCMA × CD3 BsAb produced by AbbVie (Chicago, Illinois) and is unique in its ability to maintain potent anti-MM effects while minimizing off-target toxicity and CRS, a common side effect reported with other BCMA-directed BsAb therapies. 26 In a Phase I trial of ABBV-383, 124 RRMM patients received ABBV-383 between 0.025 and 120 mg intravenously every 3 weeks. 27 Patients receiving ABBV-383 at doses ⩾40 mg achieved an ORR of 68%. For all efficacy, evaluable patients (n = 122; all doses), the ORR was 57% and ⩾VGPR rate was 43%. The most common hematologic treatment-emergent adverse events (TEAEs) were neutropenia (all grades: 37%) and anemia (29%). The most common nonhematologic TEAEs were CRS (57%) and fatigue (30%). Serious CRS events occurred in 22 (18%) patients in the overall population, 21 (26%) of which were in the ⩾40 mg escalation plus expansion cohorts. With a median follow-up of 10.8 months, the median DOR and PFS have not been reached. 27
Linvoseltamab
Linvoseltamab is a BCMA × CD3 BsAb produced by Regeneron Pharmaceuticals (Tarrytown, New York) for the treatment of RRMM patients. In recently published data from a Phase I trial, 45 patients received linvoseltamab between 3 and 96 mg intravenously over six dose levels QW followed by a maintenance dosing phase consisting of drug administration every 2 weeks.
28
Patients in this trial achieved an ORR of 35.6% among all dose levels and an ORR of 60% among patients who received the highest dose level. Of those that responded, 81.3% of patients achieved a very good partial response (VGPR) and 31.3% a CR or stringent (s)CR. Furthermore, 41.3% of responders had a DOR
Alnuctamab
Alnuctamab (CC-93269) is an anti-BCMA and anti-CD3 trivalent BsAb and has recently been evaluated in a phase I trial (NCT03486067) for 30 patients with RRMM. 30 This study included doses ranging from 0.15 to 10 mg, administered intravenously. Response rates were higher among patients receiving doses ⩾3 mg, and ORR was 36% among patients treated with the 3–6 mg dosing and 89% among those treated with >6 mg dosing. Overall, the ORR was 54% and the median time to response was 4.1 weeks (range, 4.0–13.1 weeks). In addition, 92% of the patients that responded were MRD negative. Most (97% (29/30)) patients had at least one TRAE. Twenty-two (73%) patients experienced a grade 3 or higher AE, including neutropenia (43%), anemia (37%), infections (30%), and thrombocytopenia (17%). CRS occurred in 23 patients (77%), including 1 grade 5 (death related to CRS). 29 The intravenous administration of alnuctamanb and the associated high incidence of CRS have led to efforts to investigate the safety and efficacy of subcutaneous administration. Recently published data showed that this route of administration significantly reduced all CRS events (53%) which were limited to grade 1 (45%) or grade 2 (9%), improving the safety profile of alnuctamab. 30
AMG420
AMG 420, coined a bispecific T-cell engager (BiTE) by its manufacturer Amgen (Thousand Oaks, California), is another BCMA × CD3 therapy being studied for the treatment of RRMM patients. In the Phase I trial of AMG 420 (NCT02514239), 42 RRMM patients received doses of AMG 420 between 0.2 and 800 μg/day in 6-week cycles consisting of four continuous weeks of intravenous administration followed by 2 weeks without therapy for up to 10 cycles.
30
The ORR for patients in all dose levels was 31% (10/42) but 70% (7/10) at the maximum tolerated dose (MTD) of 400 μg/day. Notably, half (21/42) of patients suffered serious (S)AEs with 18 requiring hospitalizations. Treatment-related SAEs occurring in
A more detailed list of ongoing BCMA-directed BsAbs for the treatment of MM can be found in other reviews
Antibody-drug conjugates
ADCs represent another drug class targeting BCMA for the treatment of MM.18,20,31 Agents in this drug class are designed with the intent of minimizing systemic toxicity while simultaneously enhancing the targeted destruction of MM tumor cells. 32 The ADC consists of three parts: a monoclonal antibody targeting an antigen on the tumor cell, a cytotoxic molecule, and a chemical linker that attaches the cytotoxic molecule to the antibody (Figure 2). As the antibody attaches to the antigen, in this case, BCMA, the complex is taken in by endocytosis and processed in endosomes. The cytotoxic molecules are then released into the cell which leads to apoptosis. 33 Below, we discuss several ADCs targeting BCMA that have been either approved or are currently in clinical trials.
Belantamab mafodotin (GSK2857916)
Belantamab mafodotin (GSK2857916) is a novel anti-BCMA antibody-drug conjugated to the microtubule-disrupting agent monomethyl auristatin F which was developed by GlaxoSmithKline (Brentford, United Kingdom) and previously approved for use in patients with RRMM. The DREAMM-2 trial was a randomized two-arm phase II trial investigating the safety and efficacy of belantamab mafodotin in RRMM patients. 34 This study included a heavily pretreated patient population with patients having received ⩽4 prior lines of therapy stratified into the 2.5 mg/kg cohort (median 6 prior lines of therapy) and those having received >4 prior lines of treatment categorized into the 3.4 mg/kg cohort (median 7 prior lines of therapy). The drug was administered intravenously every 3 weeks on day 1 of each cycle. The ORRs in the 2.5 and 3.4 mg/kg groups were 31% and 34%, respectively. The most common AEs were corneal events, occurring in 70.5% of the 2.5 mg/kg group and 76.8% of the 3.4 mg/kg group, and thrombocytopenia (22% in the 2.5 mg/kg dose and 33% in the 3.4 mg/kg dose). More than 40% of patients in both the dose cohorts experienced SAEs, including one death in each group. Belantamab mafodotin was removed from the US market in November of 2022, as the DREAMM-3 trial did not meet endpoints to support its continued approval. 34 One limitation of this study was that not all patients recruited were triple-class refractory.
AMG 224
AMG 224 is an anti-BCMA IgG1 antibody that is conjugated to mertansine, an anti-tubulin inhibitor. A phase I trial is currently being conducted to evaluate this agent in the setting of RRMM. 35 Forty patients were enrolled and received treatment in this trial. They had a median of 7 prior lines of therapy. AMG 224 was administered intravenously every 3 weeks to 29 and 11 patients in the dose escalation (30–250 mg) and dose expansion (3 mg/kg) phases, respectively. The ORR for all dose levels was 23% and the median DOR in the dose escalation arm was 14.7 months. In the dose escalation portion of the trial, 29 (100%) had TEAEs, with 15 patients with grade >3 AEs, the most common being thrombocytopenia (24%) and anemia (21%). Treatment-related ocular AEs (grade 1 or 2) occurred in 6 (21%) patients. SAEs occurred in 9 (31%) of patients, which included thrombocytopenia and infusion-related reactions. Notably, no patient in the dose expansion discontinued treatment due to AEs. 35
MEDI2228
MEDI2228 is an anti-BCMA ADC that utilizes a pyrrolobenzodiazepine (PBD) dimer as a toxic payload. The PBD tesirine has potent anti-tumor activity and is conjugated to the anti-BCMA mAb, BCMA-Ab-1, in MEDI2228. 36 MEDI2228 is currently being studied in a phase I trial for patients with RRMM. 37 Escalating dose levels were administered intravenously every 3 weeks. Eighty-two patients were enrolled and had previously received 2–11 prior lines of therapy. The MTD was 0.14 mg/kg and the most common TRAEs were photophobia (54%), thrombocytopenia (32%), rash (30%), increased gamma-glutamyltransferase (24%), dry eye (20%), and pleural effusion (20%). At the MTD of 0.14 mg/kg, the ORR was 61% with a median DOR that has not been reached. Preclinical studies have additionally demonstrated the ability of MEDI2228 to induce synergistic myeloma cytotoxicity with the CD38 targeting monoclonal antibody daratumumab, providing the basis for combination therapies with MEDI2228. 38
CAR-T cell therapy
CARs are reengineered proteins that can be expressed on patient-derived T-cells to enhance the ability of those T-cells to recognize and destroy cancer cells in an MHC-independent manner 39 (Figure 2). Furthermore, there is an extensive production time required for generating the cell product and a limited number of facilities with the capability for production. This limitation not only reduces their availability but also causes delays in their timely administration within the RRMM setting. These problems have severely limited the widespread use of these cell-based products. 40
To reduce the risk of off-target-related toxicities, a CAR should target an antigen that is both highly and consistently expressed in malignant cells but absent from normal cells.41,42 CD19 has been suggested to be on the myeloma stem cell by some investigators 43 ; thus, it has been used as a target for CAR-T cell therapy. However, its expression is not found in the majority of myeloma cells. 44 Thus, it was unsuccessful when CAR-T cells targeting this antigen were used in combination with autologous stem cell transplant to treat 10 patients with advanced MM. 44 This antigen is also expressed in many nonmalignant B-cell populations raising the risk of increased immunosuppression for patients treated with CAR-T cell therapies targeting this antigen. 45 CD38 has been studied in MM and is also found on activated T-cells and nonmalignant B-cells; and, therefore, targeting this antigen with CAR-T cells increases the risk for T-cell fratricide. 46 Preclinical studies have shown that CAR-T cells targeting this antigen eliminate MM cells but also eliminate nonmalignant hematopoietic cells. 47 A combination of CD38 and BCMA CAR-T cells has been evaluated in 22 RRMM patients in a phase II study with a 90.9% ORR with 54.5% achieving CR. 48 However, it is not possible to determine the contribution of the CAR-T cells targeting CD38 to the results from this dual-targeting therapeutic approach. CD138 is another possible option to target using CAR-T cell therapy. This antigen has been targeted using an ADC approach with significant off-targeting effects on the skin and liver. 49 It also is not found on all of the myeloma cells and has off-targeting effects yet to be evaluated in the clinical setting. 46 A small phase I study involving five RRMM patients treated with CAR-T cells targeting CD138 did not achieve any responses. 50 Because of the high expression of BCMA on myeloma cells, it has been considered an ideal target antigen in CAR-T therapies for MM. The first-in-human clinical trial of CAR-T cells targeting BCMA (NCT02215967) offered promising results with an ORR of 81% and 63% of patients achieved at least a VGPR. 51 In the following section, we discuss the numerous CAR-T cell therapies targeting BCMA that have followed this initial study and have been recently approved or are currently in clinical trials.
Idecatagene vicleucel
Idecatagene vicleucel was evaluated in the EVOLVE (NCT03430011), phase I/II trial assessing its safety and efficacy among patients with RRMM.52,53 Eligible patients had undergone at least three previous treatment regimens that included a PI, IMiD, and anti-CD38 antibody. A total of 140 patients were enrolled in this trial, and 128 patients received idecatagene vicleucel. At a median follow-up time of 13.3 months, 94/128 (73%) of patients had a response and 42/128 (33%) had a CR or better. Of note, MRD negative status was achieved in 33 patients (26%). Common side effects included neutropenia in 117 patients (91%), anemia in 89 (70%), and thrombocytopenia in 81 (63%). In addition, CRS was seen in 107 patients (84%) and neurotoxic effects occurred in 23 patients (18%). The results of this trial led to idecabtagene vicleucel being the first FDA-approved CAR-T cell therapy for MM in 2021.52,53
Ciltacabtagene autoleucel
CARTITUDE-1 (NCT03548201), a single-arm, open-label phase Ib/II study, aimed to assess the safety and clinical activity of ciltacabtagene autoleucel (cilta-cel) for treating RRMM patients. 54 Of the 97 patients that received cilta-cel at a dose of 0.75 × 106 CAR-positive viable cells/kg, the ORR was 97% with 65% achieving stringent CR. The observed responses were early and deep with time to first response being 1- and a 12-month PFS rate of 77%. The MRD negativity rate was 63%. Hematological AEs were common; grade 3–4 hematological AEs were neutropenia (95%), anemia (68%), leukopenia (61%), thrombocytopenia (60%), and lymphopenia (50%). CRS occurred in 95% of patients (4% were grade 3 or 4). CAR T-cell-related neurotoxicity occurred in 21% of patients (9% were grade 3 or 4). During the study, 14 deaths occurred: 6, 5, and 3 due to TRAEs, progressive disease, and 3 because of treatment-unrelated AEs, respectively. 54
Bb21217
Bb21217 is a next-generation anti-BCMA CAR-T cell therapy that is based on idecatagene vicleucel but is cultured with the phosphoinositide 3-kinase inhibitor bb007, enriching the product for more memory-like T-cells. There is evidence to suggest that CAR-T cells with this phenotype may be more persistent and more effective. 55 The safety and efficacy of Bb21217 for the treatment of RRMM are currently being evaluated in an ongoing multicenter phase I dose escalation trial (NCT03274219). 55 Initially, patients received Bb21217 at one of four dose levels (150, 450, 800, and 1200 × 106 CAR+ T-cells) with intermediate doses allowed. As the study progressed, doses were limited to either 150, 300, or 450 × 106 CAR+ T-cells. As of February 16th, 2021, 72 patients had received Bb21217. This patient population was heavily pretreated with a median of 6 prior lines of therapy and 49 patients (68%) were triple-class refractory. The ORR across the four dose levels (150, 450, 800, and 1200 × 106 CAR+ T-cells) was 69% with 20 patients achieving CR. CRS was seen in 54/72 patients (grade 1/2 in 51, grade 3 in 1, and two deaths) in the four dose levels and responded to supportive care, tocilizumab, and/or corticosteroids. Eleven patients (15%) developed neurotoxicity (grade 1 and 2 (n = 8), grade 3 (n = 2), and grade 4 (n = 1)). The median DOR across all doses was 17 months (range, 11–35 months). Overall, this specific CAR-T cell therapy showed encouraging response rates and manageable AEs consistent with known side effects for CAR-T cell therapy. 56
LCAR-B38M
A phase I, single-arm, open-label, multicenter study (NCT03090659) enrolled 57 patients with RRMM to investigate the safety and, as a secondary objective, the anti-myeloma activity of LCAR-B38M. 57 This agent is unique in that it is a dual epitope binding CAR-T cell therapy directed against two distinct epitopes of BCMA. In this patient population, the median number of prior lines of therapy was 3, 68% of patients received prior PIs, 86% received prior IMiDs, and 60% received both at least one prior PI and IMiD. The most common (⩾ 40%) AEs of any grade were pyrexia (91%), CRS (90%), thrombocytopenia (49%), and leukopenia (47%). Grade ⩾ 3 AEs were reported in 37 patients (65%); the most common (⩾ 20%) grade ⩾ 3 events were leukopenia (30%), thrombocytopenia (23%), and increase in aspartate aminotransferase levels (21%). The ORR was 88% and 39 patients (68%) achieved a CR, three patients (5%) achieved a VGPR, and eight patients (14%) achieved a PR. MRD negativity was demonstrated in 36 patients (63%). There was also a decrease in tumor mass in patients with EMD. Overall, this anti-BCMA-targeted therapy has an AE profile consistent with known toxic effects of CAR-T cell therapy and impressive initial clinical activity. 57 At the 4-year follow-up, the LEGEND-2 trial 58 further demonstrated the durable responses seen in patients with an ORR of 87.8%, and 73% of patients achieving a CR. The median DOR was 23.3 months and the median PFS was 18 months while the median OS has not been reached. 58
Serum BCMA as a biomarker
In addition to the potential of BCMA as a therapeutic target, sBCMA has also been shown to be a potential new, effective biomarker for the diagnosis, prognosis, and monitoring of MM patients. Due to the direct production of BCMA by PCs, measurement of sBCMA has been found to correlate well with disease status. In a 2012 study conducted by our group, we found that BM mononuclear cells from MM patients showed higher expression of membrane-bound BCMA versus healthy donors. 59 Furthermore, the percentage of MM plasma cells in the BM correlated with sBCMA levels in these patients, and changes in its levels correlated with changes in the clinical status of patients undergoing anti-MM therapy. 59 Taken together, measurement of sBCMA represents a valuable tool for clinicians to use to better understand patients’ disease status and response to treatment. Due to the ability of BCMA to solubilize in the blood, measurement of its serum levels provides great utility in assessing disease status in a noninvasive manner.
sBCMA as a diagnostic marker
Critical to the workup of patients with plasma cell dyscrasia is the ability to define differences between active MM patients and those with other disorders that do not require immediate treatment, specifically monoclonal gammopathy of undetermined significance (MGUS) and smoldering (S) MM. Traditionally, a BM biopsy (BMX) to define the percentage of monoclonal plasma cells, the levels of the monoclonal proteins, and the presence of hypercalcemia, anemia, kidney injury, and lytic lesions have been the gold standards used to differentiate between these disease states. 60 However, these types of diagnostic procedures especially related to the BM examination have their limitations. Specifically, underestimation of plasma cell load in the BM aspirate smear is possible due to both blood contamination and variability in the disease distribution. 60 Additionally, BM core samples have been shown to contain a greater plasma cell load than their aspirate counterparts.60–63 The combination of these factors can cause discrepancies when determining patients’ disease status. Physicians often rely on the BX in addition to disease biomarkers to fully assess patients’ tumor burdens. Biomarkers utilized to identify the presence and progression of MM have traditionally been the secreted products from malignant plasma cells and include assessment of the levels of sMPs and sFLCs.
More recently, sBCMA, the cleaved form of BCMA, has been identified as a promising biomarker with its levels correlating with patients’ disease status and being predictive of the time to disease progression. In a 2012 study, it was found that sBCMA levels measured in MGUS patients and healthy subjects were significantly lower than in untreated MM patients. 59 In a 2017 study, healthy donors were found to have a median sBCMA of 36.8 ng/mL, smoldering MM patients of 88.9 ng/mL, and active untreated MM patients of 505.9 ng/mL. 64 Thus, levels of sBCMA in addition to sMP and sFLC appeared to correlate with the severity of the plasma cell dyscrasia.
sBCMA as a prognostic marker
In addition to the potential of sBCMA as a diagnostic marker, it has also shown promise as a way to predict treatment and survival outcomes among patients with MGUS, SMM, and active MM. In a 2021 study, baseline sBCMA was measured in 65 SMM patients, and a level of 137.5 ng/mL was determined to be the threshold, with levels below being predictive of low risk and levels above being predictive of high risk for disease progression to active MM. 65 Of those in the high-risk category, 42.9% of patients transformed to active MM whereas only 7.7% in the low-risk group did. Furthermore, patients stratified in the high-risk group were also found to have a shorter time to transformation. Of note, sBCMA was independent of other high-risk factors and was the only variable found to be significantly predictive of time to transformation. In a separate study. Visram et al. 66 evaluated outcomes in 99 MGUS and 184 SMM patients who were categorized as high risk if their sBCMA levels were ⩾77 and⩾128 ng/mL, respectively. High-risk MGUS patients were found to have a PFS of 3.9 years versus 11.5 years among those in the low-risk group. Similarly, high-risk SMM patients showed a median PFS of 1.9 years versus 4.7 years among patients with low-risk disease. There was also a longitudinal increase in sBCMA among patients with MGUS and SMM who progressed to active MM. Among those who progressed to MM, the median sBCMA increased by 2.7-fold in MGUS patients and 1.3-fold in patients with SMM whereas there were no significant changes among those who did not progress. Interestingly, there was no significant difference between the fold change of MGUS and SMM patients that progressed. 66
sBCMA as a monitoring tool
Changes in sBCMA have been shown to serve as a reliable predictor of changes in clinical status and OS among MM patients starting a new treatment. First, Jew et al.67,68 determined levels among healthy subjects, and the median sBCMA level was found to be 37.51 ng/mL with the upper threshold of normal to be 82.59 ng/mL. MM patients who started a new treatment with a sBCMA in the normal range (i.e., <82.59 ng/mL) had improved PFS and OS. Furthermore, among patients who began MM therapy with sBCMA levels above the normal range and subsequently decreased to normal range after treatment demonstrated an improved OS. The same study showed that patients whose sBCMA normalized also demonstrated improved ORR and all those patients who achieved a CR showed normalization of sBCMA. Of note, time to normalization of sBCMA was faster than time to CR. 67 Bujarski et al. 69 recently reported on 81 RRMM patients starting a new treatment and found that their median sBCMA level was 305.5 ng/mL. Patients above the median level were found to have a shorter PFS (median 2.6 months) than individuals with amounts below the median (median 8.0 months). Furthermore, those in the highest quartile (⩾594.8 ng/mL, n = 20) were found to have much shorter median PFS (1.8 months) versus the other three quartiles (7.3 months; n = 61; p = 0.0012). The same study showed that patients with a ⩾25% increase in sBCMA between weeks 4 and 12 of treatment had a shorter PFS (median 1.8 months) versus those with <25% increase (median 6.8 months). Of note, a ⩾25% increase in sBCMA in 67.5% of patients before disease progression occurred based on International Working Group Criteria (median, 13 days faster). Additionally, those with a ⩾50% decrease in sBCMA between weeks 4 and 12 of therapy had a longer PFS (median 7.8 months) versus those who did not meet this decline in their sBCMA level (median 2.7 months). 69 Overall, these findings provide support for this new biomarker to provide a new and noninvasive way to longitudinally assess patients with MM.
Comparing sBCMA to other MM biomarkers
As a biomarker, sBCMA holds several advantages over traditional biomarkers used to monitor MM patients, specifically sMP and sFLC. Among patients with secretory disease, sBCMA was found to have a more rapid turnover rate (24–36 h) compared to sMP (3–4 weeks) and it was shown that monitoring sBCMA weekly for the first cycle of new treatment was quicker to define changes in clinical status than monitoring sMP due to the more rapid turnover rate.59,70 Furthermore, unlike the levels of sFLC which have been found to be 20- to 30-fold higher among patients with renal failure, sBCMA levels were found to be independent of renal function making it a more useful biomarker for MM patients who often experience renal impairment from their disease and other disease and age-related problems. 71 Additionally, sBCMA has been shown to be the first circulating biomarker that can effectively track patients with nonsecretory MM which is characterized by the absence of detectable M-protein or abnormal FLCs in the serum and urine.60,64 This gives clinicians the ability to monitor the course of disease among nonsecretory MM patients noninvasively without the use of frequent BMXs and expensive imaging procedures such as positron emission tomography CT scans.
Challenges facing BCMA-directed therapies
Although BCMA-directed MM therapies represent a promising future for MM patients, there are several challenges still facing these treatment options, particularly in their efficacy among patients with advanced disease and the frequent occurrence of severe TRAEs. When BCMA is cleaved from the cell surface by gamma secretase, it becomes solubilized in the blood where high concentrations of the protein fragment may accumulate; and, thus, it may bind to therapeutic anti-BCMA antibodies, inhibiting their effectiveness. In an in vitro study, Chen et al. 72 showed that an anti-BCMA antibody showed consistently decreased binding to malignant plasma cells when the cells were cultured with serum from MM patients whose sBCMA levels were ⩾156 ng/mL. In the same study, the authors reported the median sBCMA levels of RRMM patients was 176 ng/mL when starting a new therapy. 72 Taken together, it is possible that many patients with elevated sBCMA levels due to advanced MM may be less responsive to BCMA-targeted immune-based therapies due to the presence of circulating BCMA. There are possible solutions, however, to prevent the shedding of BCMA that may improve BCMA-directed therapies. In a 2019 study, 73 it was shown that gamma-secretase inhibitors (GSIs) increased surface BCMA and decreased sBCMA which ultimately improved efficacy of BCMA CAR T-cell therapy in MM tumor-bearing mice. It is possible that the concomitant use of GSIs with other anti-BCMA-directed therapies may prove to be a viable option for patients with advanced disease. In addition, several tumor-intrinsic factors have recently been identified in facilitating BCMA antigenic escape. A 2023 study reported that among 30 patients treated with anti-BCMA CAR-T/TCE (T-cell engager) therapy, MM relapse post-treatment in two patients was attributed to the expansion of BCMA-negative clones while relapse in five patients was attributed to mutations within the extracellular domain of BCMA. 74 This highlights the ability of antigenic escape to occur despite maintained expression of BCMA via loss of a functional epitope in the case of mutations within the extracellular domain. Continuing to develop a deeper understanding of the diverse and complex mechanisms by which antigenic escape occurs will positively alter the design and implementation of these types of immunotherapies.
Regarding patient safety while utilizing anti-BCMA-directed therapies, advancements must be made to allow for their sustained use. Some ADCs especially belantamab mafodotin have been associated with a significant amount of ocular toxicity. In the expansion portion of the phase I trial of this ADC, 63% of patients reported corneal events with a median time to onset of 23 days. 75 In addition, CAR-T cell therapies have also been shown to have significant dose-limiting toxicities with the most concerning being CRS and ICANS. Life-threatening complications associated with these syndromes include cardiac dysfunction, adult respiratory distress syndrome, neurologic toxicity, renal and hepatic failure, and disseminated intravascular coagulation. 76 Glucocorticoids, known for their ability to suppress inflammatory reactions, have been recently utilized to mitigate the risk of developing these syndromes and for the management of symptoms once these syndromes have appeared. Additionally, more specific approaches involving the blockade of subgroups of cytokines using antibodies, especially the anti-IL-6 antibody tocilizumab, and kinase inhibitors have been utilized in this setting.77–79 The advancement of concomitant prophylactic therapies will improve the clinical safety profile for CAR-T therapies and expand their usage among diverse patient populations.
The CAR T-cell-related hematologic toxcicity (CARHEMATOX) score was originally designed to risk stratify patients with large B-cell lymphoma for toxicity events as well as clinical outcomes prior to receiving CAR-T cell therapy. A study by Rejeski et al. also validated this score among patients receiving BCMA-directed CAR-T cell therapies. The score incorporates factors related to hematopoietic reserve prior to CAR-T cell therapy, such as hemoglobin, absolute neutrophil count, and platelet count as well as inflammatory markers such as ferritin and C-reactive protein. 80 The results of this study showed that a high CARHEMATOX score (>1) versus a low score (0–1) was associated with prolonged severe neutropenia, an increase in severe infections, and more ICANS. Response rates were also higher in patients with a low score versus a high score. 80
Neurological complications are also another complication secondary to BCMA-directed CAR-T cell therapy. One such complication, as mentioned prior, is ICANS. The reasons why this problem occurs are not well understood but are thought to be related to increased cerebrospinal fluid cytokine levels and disruption of the blood-brain barrier. 81 It is characterized by neurological dysfunction that can range from headaches, fatigue, and tremors, to coma and even death. It usually develops approximately 3–10 days following administration of the CAR-T cell product. Grading is through the 10-point Immune Effector Cell-Associated Encephalopathy score. Dexamethasone is usually administered in moderate to severe ICANS (grade >1). Given the concomitant development of CRS, patients usually receive tocilizumab as well. 82 Another neurologic event with BCMA-directed CAR-T cell therapies that resembles parkinsonism has been reported in the literature. 83 These cells can cross the blood-brain barrier in select patient group and through targeting BCMA expressing basal ganglia cells can cause a progressive neurocognitive and hypokinetic movement disorder. 83
Infectious complications with BCMA-directed therapies have been well documented in the literature. One study by Sim et al. determined infectious complications among MM patients receiving bispecific BCMA-targeted antibodies. Among the 39 patients in their institution treated with these agents, 35 (90%) had at least one infection. They identified a total of 111 infections, with the most common site being respiratory (41%) followed by gastrointestinal (7%). The most common viral infection was rhinovirus/enterovirus, followed by cytomegalovirus, and adenovirus. The majority of bacterial infections were gastrointestinal and there were no episodes of invasive fungal disease. There may be a role for Pneumocystis prophylaxis with sulfamethoxazole/trimethoprim and herpes/zoster prophylaxis with valaciclovir or similar drugs. Fungal prophylaxis should be guided by individual risk. 84
Mechanisms of resistance of BCMA therapy
The mechanism of resistance to BCMA-directed therapies is still being investigated and as of now is poorly understood. One thought regarding resistance is biallelic or monoallelic loss of BCMA on chromosome 16 as well as point BCMA mutations. 85 Downregulation of its expression may occur in the absence of mutation of the gene. Another proposed mechanism of resistance includes sBCMA released through the activity of gamma secretase which then acts as a decoy receptor for BCMA-directed therapies. 18 In addition, T-cell exhaustion especially in the setting of BsAbs has been another proposed mechanism of resistance. 86
Conclusion
The specificity and abundance with which BCMA is expressed on malignant plasma cells makes it an ideal therapeutic target for treating MM patients. This promising potential has led to the development of novel BCMA-focused therapies, including BsAbs, BiTEs, ADCs, and CAR-T cell therapies. There are currently several FDA-approved therapies targeting this protein for the treatment of MM including the ADC belantamab mafodotin, the BsAbs teclistamab and elranatamab, and the CAR-T’s idecatagene vicleucel and cilta-cel. These agents have shown a great deal of promise as shown by the MRD negativity rates achieved in the clinical trials mentioned above (Table 1). MRD is a valuable tool as it correlates with both PFS and OS and may become a useful tool to help guide treatment decisions in the near future.87–89 While severe TRAEs have been associated with BsAb and CAR-T-based therapies such as CRS and ICANS, these treatments demonstrate exceptionally high and durable rates of clinical response. As basic and clinical research progresses, the safety and efficacy of this class of therapeutics will ideally improve, allowing the widespread use of these therapies in a more diverse patient population. Additionally, the cleavage of BCMA by gamma secretase to produce a serum soluble protein fragment, sBCMA, presents an opportunity to exploit this protein as a blood biomarker for characterizing plasma cell dyscrasias and improving monitoring for patients with these disorders including those lacking conventional markers to follow their disease.
MRD negative rates for BCMA-directed therapies.

Supplemental Material
sj-docx-1-tah-10.1177_20406207241275797 – Supplemental material for Targeting B-cell maturation antigen for treatment and monitoring of relapsed/refractory multiple myeloma patients: a comprehensive review
Supplemental material, sj-docx-1-tah-10.1177_20406207241275797 for Targeting B-cell maturation antigen for treatment and monitoring of relapsed/refractory multiple myeloma patients: a comprehensive review by David Yashar, Bernard Regidor, Marissa-Skye Goldwater, Sean Bujarski, Ashley Del Dosso and James R. Berenson in Therapeutic Advances in Hematology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.