
Abstract
The treatment of multiple myeloma (MM) has evolved substantially over the past decades, leading to a significantly improved outcome of MM patients. The introduction of high-dose therapy, especially, and autologous stem cell transplantation, as well as the development of new drugs, such as immunomodulatory drugs (IMiDs) and proteasome inhibitors have contributed to the improvement in survival. However, eventually most MM patients relapse, which indicates that there is a need for new agents and novel treatment strategies. Importantly, the long-term survival in a subset of MM patients after allogeneic stem cell transplantation illustrates the potential of immunotherapy in MM, but allogeneic stem cell transplantation is also associated with a high rate of treatment-related mortality. Recently, a better insight into several immune-evasion mechanisms, which contribute to tumor progression, has resulted in the development of active and well-tolerated novel forms of immunotherapy. These immunotherapeutic agents can be used as monotherapy, or, even more successfully, in combination with other established anti-MM agents to further improve depth and duration of response by preventing the outgrowth of resistant clones. This review will discuss the mechanisms used by MM cells to evade the immune system, and also provide an overview of currently approved immunotherapeutic drugs, such as IMiDs (e.g. lenalidomide and pomalidomide) and monoclonal antibodies that target cell surface antigens present on the MM cell (e.g. elotuzumab and daratumumab), as well as novel immunotherapies (e.g. chimeric antigen receptor T-cells, bispecific antibodies and checkpoint inhibitors) currently in clinical development in MM.
Keywords
Multiple myeloma
Multiple myeloma (MM) is a malignant disease, characterized by clonal proliferation of plasma cells in the bone marrow. It is a disease of the elderly, with a median age at diagnosis of 70 years and only 5% of patients below 40 years. MM accounts for 1% of all malignancies and approximately 10% of all hematological malignancies. The disease originates from a, generally asymptomatic precursor disease, monoclonal gammopathy of undetermined significance (MGUS). The process of transformation of MGUS to symptomatic MM is associated with sequential genetic and micro-environmental changes. 1 Clinical characteristics of MM include osteolytic bone lesions, hypercalcemia, renal failure, and progressive bone marrow dysfunction with anemia and other cytopenias.1,2 The treatment of MM has undergone significant changes and improvements in recent years. Next to the development of proteasome inhibitors (such as bortezomib, carfilzomib and ixazomib), the introduction of immunomodulatory drugs (IMiDs) that have profound stimulatory effects on the immune system and direct anti-MM activity (such as thalidomide, lenalidomide and pomalidomide), as well as the development of novel immunotherapies has significantly improved the outcome of MM patients over the past decade.3,4 However, even though median survival currently is 7–10 years, eventually most patients relapse. This indicates the need for new anti-MM agents or potentiation of the currently available therapies by development of alternative treatment combinations and strategies. Also increased insight into resistance mechanisms to current treatments may lead to new strategies to prevent or overcome treatment resistance. This review aims to provide an outline of the mechanisms of immune evasion in MM, which is followed by a discussion of currently approved immunotherapy drugs, as well as novel immunotherapies in various stages of clinical development in MM.
Immune evasion by MM cells
Immunotherapy can induce potent clinical responses in MM, as is illustrated by long-term survival in a subset of patients after allogeneic stem cell transplantation (allo-SCT) and the successful results of the immunotherapeutic approaches that will be described below. However, like other malignancies, MM cells have several mechanisms to escape immune-mediated killing. Insight into these mechanisms may lead to potential interventions to improve immune therapy in MM. In the following section, a number of mechanisms of immune evasion by MM cells are described.
Immune suppressor cells
The two most studied immunosuppressive cell subsets that can be induced by MM cells are regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSCs). Regulatory T-cells are a subset of T-cells characterized by the expression CD4, CD25, and forkhead box P3 (FOXP3) and low expression of CD127 (CD4+CD25+CD127lowFOXP3+). Tregs can be naturally occurring (generated in the thymus) or induced from effector T-cells, and are biologically important for maintaining peripheral tolerance and limiting auto-immune disease. However, they can also suppress anticancer immune responses. Tregs exert their suppressive effect through several mechanisms. They secrete suppressive cytokines, such as transforming growth factor (TGF)-β and IL-10, and have the capacity to kill B-cells, natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), by secreting granzyme-B. In addition, Tregs suppress the function of dendritic cells through interaction of CTLA-4 and lymphocyte activation gene 3 (LAG3) with their ligands on the dendritic cells (DCs). Furthermore, Tregs induce expression of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO) by DCs. 5 Several reports show increased frequencies of Tregs in peripheral blood of MM patients.6–10 Furthermore, MM cells were shown to induce Tregs from conventional T-cells ex vivo.11,12 However, others state that these Tregs are dysfunctional, or show similar levels of Tregs in MM patients compared with healthy donors.13–16 These conflicting results can in part be explained by (1) differences in definition of the Treg phenotype, (2) compartment where Tregs are measured (peripheral blood versus bone marrow) and (3) disease status (newly diagnosed versus relapsed/refractory MM). In line with the idea of MM-induced Treg expansion and active immune suppression are two studies, which show that lower Treg numbers in bone marrow and peripheral blood are associated with long-term survival in MM patients.17,18 Furthermore, recent reports show an increased CD38 expression on Tregs as compared with conventional T-cells, whereby alleviation of Treg-induced immune suppression in MM can be achieved using CD38-targeting antibodies such as daratumumab and isatuximab.12,13,19
MDSCs are a heterogeneous, immature population of CD11b+CD33+HLA-DR-/low myeloid cells. Two main subtypes of MDSCs exist: polymorphonuclear (granulocytic) MDSCs, expressing CD15 or CD66b, and monocytic MDSCs expressing CD14, both in addition to the phenotype mentioned above. MDSCs exert their suppressive function through several distinct mechanisms. They deplete essential amino acids like L-arginine and L-cysteine, and cause oxidative stress by production of reactive oxygen species and reactive nitrogen species, both inhibiting T-cell function. Furthermore, they interfere with lymphocyte trafficking and viability, and induce Tregs. 20 MDSCs have been found at increased frequencies in peripheral blood and bone marrow of MM patients, compared with healthy donors.21–25 In addition, MM cells were shown to induce MDSCs, and conversely, MDSCs contributed to disease progression in MM. 24 These results indicate an active immunosuppressive and disease-promoting role of MDSCs in MM.
In addition to Tregs and MDSCs, regulatory B-cells (Bregs) have been described to play a role in MM. Bregs are a subset of B-cells identified by the CD19+CD24highCD38high cell surface phenotype, which can regulate immune responses by production of the anti-inflammatory cytokine interleukin (IL)-10 (among other mechanisms). 26 In MM patients, Bregs were shown to be a distinct population in the bone marrow microenvironment, dependent on the presence of MM cells, and capable of suppressing anti-MM cell antibody-dependent cellular cytotoxicity (ADCC) by NK cells. 27
Growth factors and cytokines contribute to immune suppression in the MM bone marrow microenvironment
The MM microenvironment is characterized by production of several immunosuppressive cytokines. A key cytokine in pathogenesis and disease progression of MM is IL-6, produced by bone marrow stromal cells (BMSCs) and MM cells, which can inhibit NK cell function. 28 Furthermore, TGF-β production by MM cells, stromal cells and osteoblasts inhibits T-cells, NK cells and DCs.29,30
A proliferation inducing ligand (APRIL) is a ligand of B-cell maturation antigen (BCMA), primarily secreted by myeloid cells and osteoclasts, and critical for plasma cell growth and survival. APRIL was shown to upregulate genes involved in immunosuppression in MM cells [TGF-β, IL-10, programmed death ligand 1 (PD-L1)], which could be abrogated by anti-APRIL antibodies. 31 APRIL also binds to ‘transmembrane activator and calcium modulator and cyclophilin ligand interactor’ (TACI). TACI is expressed on plasma cells at a lower level as compared with BCMA. TACI is also expressed at significantly higher levels on Tregs as compared with conventional T-cells, and APRIL was shown to promote Treg viability through inhibiting apoptosis, which was abrogated by addition of anti-APRIL but also by anti-TACI antibodies. 32 APRIL also enhanced Treg-mediated inhibition of conventional T-cell proliferation, and increased the induction of Tregs by MM cells. 32
Co-inhibitory molecules
Activated T-cells express several co-inhibitory molecules (immune-checkpoint molecules) such as cytotoxic T lymphocyte associated antigen-4 (CTLA-4) and programmed death-1 (PD-1). Binding of these receptors to their corresponding ligands [CD80/86 for CTLA-4 and PD-ligand-1/2 (PD-L1/PD-L2) for PD-1] on antigen-presenting cells (APCs) leads to a controlled inhibition of activated T-cells, which confers protection against immune-mediated diseases. However, soon after the discovery of these natural protection mechanisms, it appeared that tumor cells effectively exploited such feedback loops by upregulating the expression of these co-inhibitory receptors. 33 A number of co-inhibitory molecules are described to be upregulated in MM. PD-L1 expression is increased on MM cells, which can be partly explained by interferon-γ produced by BMSCs and has been shown to induce apoptosis and anergy of myeloma-specific T-cells.34–36 In addition, PD-L1 expression on MM cells has intrinsic effects. It has been shown that PD-L1-positive MM cell lines have a proliferation advantage, increased levels of anti-apoptotic proteins, and decreased sensitivity to dexamethasone and melphalan.36,37 Besides PD-L1, MM cells express increased levels of carcinoembryonic antigen-related cell adhesion molecules (CEACAMs). CEACAM-6 has been shown to inhibit anti-MM T-cell activity, which was completely abrogated by using anti-CEACAM-6 antibodies. 38 Furthermore, a higher expression of the transmembrane glycoprotein CD200 on MM cells at diagnosis correlates with decreased event-free survival after high-dose therapy and autologous transplantation, possibly by inhibition of T-cell activity after binding to its ligand CD200R on T-cells. 39 On the T-cell side, increased expression of PD-1 and CTLA-4 was observed on bone marrow-localized T-cells of MM patients, indicating an exhausted phenotype which might be reversed by blocking these receptors with therapeutic antibodies targeting PD-1 or PD-L1. 40
Cell adhesion-mediated immune resistance
BMSCs as well as vascular endothelial cells in MM patients can suppress CTLs and NK cell-mediated killing of MM cells by regulating anti- and pro-apoptotic pathways in the MM cells.41,42 This immune resistance was shown to be induced by cell–cell adhesion and decreased significantly when adhesion was abrogated. Upon cell–cell adhesion, downregulation of Fas and an upregulation of survivin and Mcl-1 has been observed. Using the small molecule YM155, survivin and Mcl-1 could be suppressed, leading to an increased MM cell lysis by CTLs.41,42
Other mechanisms of immune suppression
Expression of indoleamine 2,3-dioxygenase (IDO) in MM cells catalyzes the metabolization of tryptophan, an amino acid essential for cell growth and functioning of immune cells, thereby leading to immune suppression. In addition, it also leads to recruitment of Tregs. 43 Increased production of soluble major histocompatibility complex class I-related chain A (sMICA) in MM leads to impaired T-cell and NK cell function. 44 In addition, studies have also shown a defective antigen-processing machinery and human leukocyte antigen (HLA) class I downregulation in MM cells. This leads to decreased recognition of the tumor cells by CD8+ T-cells.45,46
Immunotherapy in MM
The general principle of cancer immunotherapy is to administer or activate immune cells, in order to gain a more targeted approach towards cancer, thereby reducing off target side effects. This approach is in contrast with traditional chemotherapeutic treatment modalities, which are generally nonspecific cytotoxic therapies with substantial side effects. In MM, some immunotherapies are currently approved for the treatment of MM, while several other immunotherapeutic approaches are currently focus of active investigations.47–49 Here, we will discuss a number of immunotherapeutic strategies currently used outside clinical trials, or investigated in MM patients (Figure 1).

Schematic overview of immunotherapeutic options in multiple myeloma.
Allogeneic stem cell transplantation
Allo-SCT can be considered one of the first immunotherapies applied in MM. In allo-SCT, stem cells of a donor are administered to a patient, who has been pretreated with radiation and chemotherapy, or chemotherapy alone. T-cells of the donor, co-administered with the stem cells, can recognize and eliminate tumor cells resulting in the so called ‘graft-versus-tumor effect (GVT)’. Unfortunately, donor T-cells can also recognize normal tissue as foreign, thereby causing the detrimental graft-versus-host-disease (GVHD), a major cause of treatment-related mortality (TRM). In an HLA-matched setting, the GVT effect is mainly mediated by the recognition of minor histocompatibility antigens (mHags) presented on malignant cells. 50 These transplantation antigens are peptides derived from the polymorphic regions of intracellular proteins. Upon binding to HLA molecules, they are presented to the T-cells of the donor. Variations in intracellular proteins that exist between the donor and recipient are due to the evolutionary occurrence of, for instance, single nucleotide polymorphisms (SNPs) in the coding regions of the genome, creating peptides which differ in amino acid sequence. Therefore mHags are foreign antigens for the donor and can therefore induce potent allo-immune T-cell responses, even in the setting of HLA-matched transplantation (Figure 2).51,52
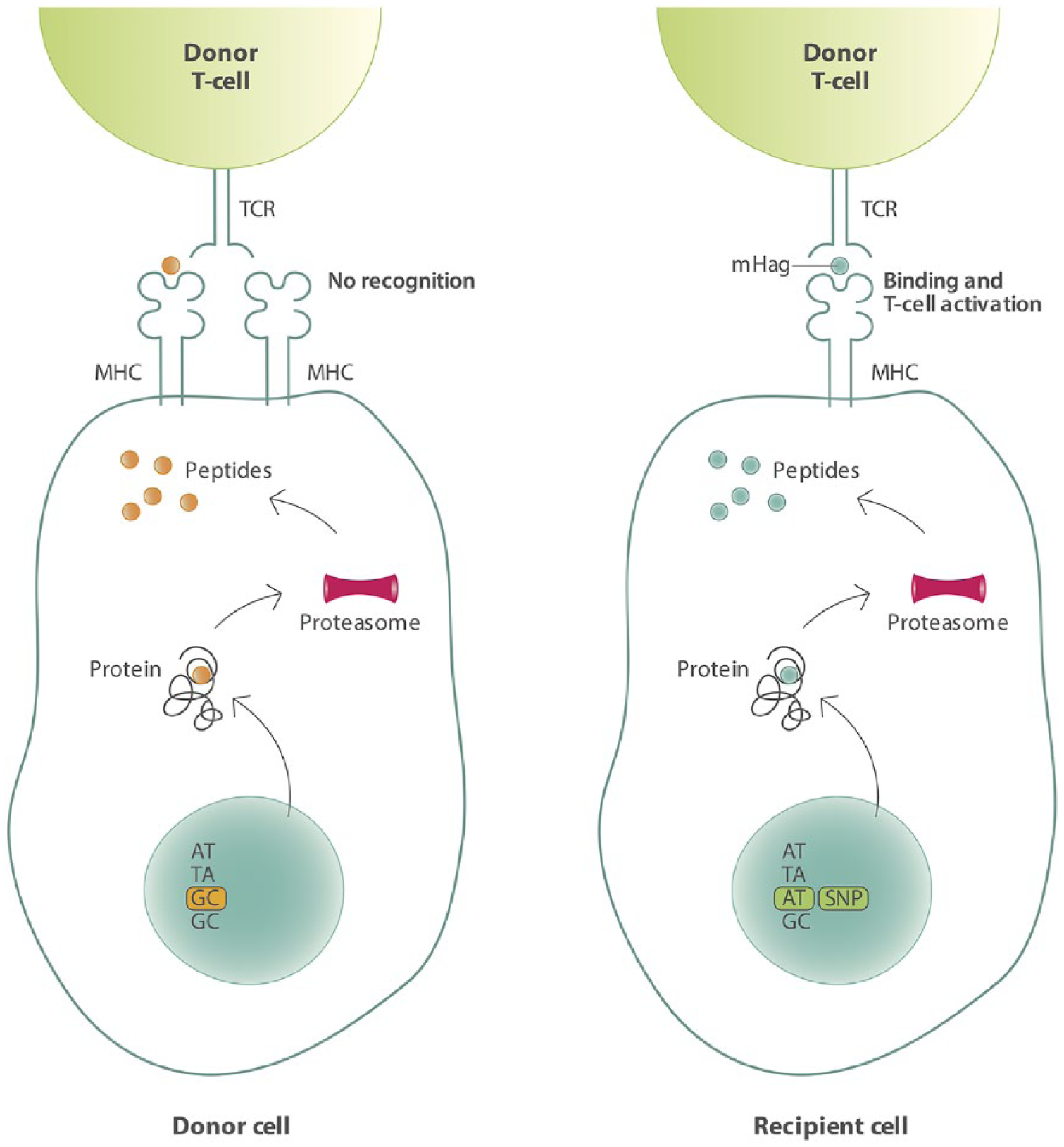
Schematic overview of mHag presentation on recipient cells leading to activation of donor T-cells.
The tissue distribution of the mHags determines their contribution to either GVT or GVHD effects. mHags expressed exclusively on hematopoietic cells have a dominant role in the beneficial GVT effect, whereas mHags with a broad tissue expression cause both GVT and GVHD. In addition to the tissue distribution, the population frequency of the mHag is important. mHags with a very low or very high population frequency are not very relevant from an immunotherapeutic point of view, as chances of a mismatch between donor and recipient will be low. The chance for a mismatch is actually maximum with an mHag population frequency of 50%.53,54
The existence of the GVT effect in MM has been clearly demonstrated in studies evaluating donor lymphocyte infusions for the treatment of relapsed MM after allo-SCT.55–57 However, although allo-transplantation may potentially induce a powerful GVT effect, which does not occur with an autologous transplantation (auto-SCT), studies comparing tandem auto-SCT versus auto-SCT followed by allo-SCT in the upfront setting have shown conflicting results.58–69 Several studies show an event-free survival and overall survival (OS) benefit with allo-SCT,58,60,61,67 while others show no difference in survival.70–75 Some of these studies suggest that allo-SCT can overcome the unfavorable prognosis in patients with adverse cytogenetic aberrations,59,76 while others found no difference in outcome in high-risk patients with MM, when double auto-SCT was compared with tandem auto-allo-SCT.66,69 However, the majority of these studies was performed before the introduction of proteasome inhibitors and immunomodulatory agents and also the definition of high-risk cytogenetics was not similar to what is currently used. This, together with a high rate of TRM of 10–20% has led to reduced application of allo-SCT as part of upfront therapy in MM. Patients with an early relapse (⩽18 months) after auto-SCT have a poor prognosis, and therefore allo-SCT can be considered in these patients, but only after successful re-induction therapy, and preferentially in the context of a clinical trial.77–80 However, the outcome of allo-SCT in these patients is not well established. Key to improvement of allo-SCT in MM patients is the development of less toxic conditioning regiments, improvement of (sustained) GVT responses, and a reduction or better treatment of GVHD. Post-allo immunotherapy modalities, such as donor lymphocyte infusions, DC vaccinations and NK cell therapy, are also the subject of investigation.81–85 It is expected that application of allo-SCT in MM will further decrease due to the introduction of other active, and less toxic, immunotherapies.
Immunomodulatory drugs
Although currently named ‘IMiDs’, and broadly applied in the treatment of MM, the IMiDs were initially not designed as such, but thalidomide (the first-in-class IMiD) was developed as a sedative, anti-emetic drug. IMiDs have multiple mechanisms of action including immune-stimulatory and anti-angiogenic properties, as well as direct anti-MM activity. 86 IMiDs bind to Cereblon, a substrate receptor for the ubiquitin E3-ligase complex CRL4CRBN, which include damage-specific DNA binding protein (DDB1), cullin 4A (Cul4) and RING finger protein 1 (ROC1).87–90 After binding, the specific substrate proteins IKZF1 (Ikaros) and IKZF3 (Aiolos) are recruited to the E3 ligase and targeted for ubiquitination and subsequent proteasomal degradation. The degradation of Ikaros and Aiolos is followed by downregulation of interferon regulatory factor 4 (IRF-4) and c-Myc leading to growth inhibition and apoptosis of MM cells (Figure 3). Ikaros and Aiolos have also been shown to act as repressors of IL-2 transcription in CD4+ and CD8+ T-cells.88,91–94 This explains that in immune cells, the IMiD-induced, Cereblon-dependent, degradation of Ikaros and Aiolos results in increased IL-2 production, while also enhancing the production of other cytokines [interferon (IFN)γ, IL-4, IL-6, IL10, IL13 and granulocyte-macrophage colony-stimulating factor (GM-CSF)]. 94 This leads to activation of T-cells and NK cells.95,96 In addition to the effect of IMiDs on T-cells and NK cells, lenalidomide has been shown to increase the expression of DC maturation markers (CD86, HLA-DR and CD209), and treatment of DCs with lenalidomide in vitro enhanced their ability to stimulate autologous T-cells.97,98 In addition, lenalidomide abrogated the inhibitory effect of mesenchymal stromal cells on DC maturation in vitro, by downregulating another Cereblon substrate protein: CK1-α. 97

The ubiquitin E3-ligase complex CRL4CRBN causing ubiquitination of IKZF1 and IKZF3 after IMiD binding, leading to their proteasomal degradation and subsequent immunomodulatory and MM cytotoxic effects.
After the recognition of the teratogenic effects of thalidomide, the drug was withdrawn from the market in 1961. In 1990, the anti-myeloma effects of thalidomide were observed after administration of the drug to five patients with end-stage MM, which led to a rapid increase in clinical trials investigating thalidomide in MM. 99 In order to improve the activity and reduce the toxicity, several new IMiDs were designed and evaluated in MM. Lenalidomide is the second-generation IMiD, which was approved in 2006 for the treatment of relapsed/refractory myeloma in combination with dexamethasone. This combination showed a significantly better response rate compared with thalidomide-dexamethasone, and also showed activity in patients previously treated with thalidomide.100,101 Importantly, lenalidomide induced significantly less neuropathy when compared with thalidomide. The third-generation IMiD, pomalidomide, was approved for treatment of relapsed/refractory MM patients who have received at least two prior lines of therapy including lenalidomide and bortezomib. Pomalidomide also has activity in lenalidomide-refractory patients, with overall response rates (ORRs) of 34% in these patients. 102 In the relapsed/refractory setting, IMiDs are used in several treatment combinations. 77 Interestingly, at this moment, several new IMiDs, such as CC220, are being investigated in phase I clinical trials in extensively pretreated MM patients. Because IMiDs showed marked anti-MM activity in relapsed/refractory MM, they were also evaluated in newly diagnosed MM patients. At this moment, lenalidomide in combination with dexamethasone is one of the standards of care for newly diagnosed MM patients who are not eligible for transplant. 77 Furthermore, in transplant eligible patients, triplets of bortezomib-dexamethasone and an IMiD [lenalidomide (VRD) or thalidomide (VTD)] are frequently used as induction regiments prior to high-dose therapy and auto-SCT. Lenalidomide also improves progression-free survival (PFS) and OS when given as maintenance therapy after auto-SCT in newly diagnosed MM patients is approved as maintenance drug in this setting. 103
Monoclonal antibodies
The substantial therapeutic effect of anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoma indicates the potential of monoclonal antibodies in anticancer treatment. 104 In contrast with the genetic heterogeneity of MM, with sequential genetic changes while the disease is progressing, the immune phenotype is more homogeneous. Furthermore, the different mode of action of immunotherapeutic treatment strategies makes cross resistance with other anti-MM drugs less likely. Identification of proteins that are highly and stably expressed on MM cells during different disease stages are attractive targets for therapeutic monoclonal antibodies.
The two antibody targets, signaling lymphocytic activation molecule F7 (SLAMF7) and CD38, are of particular interest in MM. Elotuzumab, a humanized immunoglobulin (Ig)G1 monoclonal antibody targets SLAMF7. SLAMF7 is highly expressed on plasma cells and NK cells, and to a lesser extent on a subset of activated T-cells and B-cells. Elotuzumab was shown to act primarily via ADCC, but has no single-agent activity in extensively pretreated MM. 105 Preclinical studies showed that lenalidomide potentiates elotuzumab-mediated ADCC by improving NK cell activity. 106 In a randomized phase III trial, elotuzumab (Elo), lenalidomide and dexamethasone (Rd) was compared with Rd in relapsed or refractory (94% lenalidomide-naïve) MM patients. ORRs were 79% versus 66% (p< 0.001), with a median PFS of 19.4 months versus 14.9 months respectively (hazard ratio 0.70, p< 0.001). 107 The 1, 2 and 3-year OS rates for Elo-RD were 91%, 73% and 60%, while these were 83%, 69% and 53% for Rd-treated patients, respectively. 108 Most common grade 3–4 adverse events were neutropenia and thrombocytopenia. Infusion-related reactions (IRRs) occurred in 10% of patients. Elotuzumab has also been combined with bortezomib and dexamethasone in a randomized phase II trial, and compared with bortezomib-dexamethasone, resulting in superior PFS (hazard ratio, 0.72; median PFS 9.7 versus 6.9 months). 109
The second important antibody target in MM is CD38. CD38 is highly expressed on plasma cells, followed by NK cells and subpopulations of T-cells and B-cells. Furthermore, it is also expressed on myeloid cells, erythrocytes and platelets. Several CD38-targeting antibodies have been developed (daratumumab, isatuximab, and MOR202). These antibodies have multiple mechanisms of action, including complement-dependent cytotoxicity (CDC), ADCC and antibody-dependent cellular phagocytosis. 110 Interestingly, daratumumab also reduces CD38+ immune suppressor cell populations such as Tregs, Bregs and MDSCs, potentially leading to a better anti-tumor immune response. 19 Daratumumab has potent single-agent activity, as was shown by the GEN501 and Sirius trials published in 2015 and 2016.111,112 Pooled analysis of these studies showed an ORR of 31.1%, a PFS of 4 months and an OS of 20.1 months in relapsed/refractory MM patients with a median of five prior lines of therapy. 113 Daratumumab was well tolerated. IRRs were observed in 48% of patients and consisted of chills, nausea and respiratory conditions, but only 2.7% of patients had ⩾ grade 3 IRRs. 113 The main hematological adverse events were anemia, thrombocytopenia and neutropenia. Most common nonhematological adverse events were fatigue, nausea and back pain. Based on the high activity of daratumumab in MM, other CD38-targeting antibodies were developed including MOR202 and isatuximab, which have comparable single-agent activity in advanced MM.114,115 Similar to elotuzumab, combining daratumumab with lenalidomide increased NK cell-mediated ADCC in preclinical studies, which led to several clinical trials combining anti-CD38 antibodies with an IMiD, showing significantly improved outcome of the combination when compared with anti-CD38 antibody monotherapy. In the POLLUX trial, Rd was compared with daratumumab-Rd in patients with at least one prior therapy but not lenalidomide-refractory disease. PFS at 12 months was 60.1% for Rd versus 83.2% for daratumumab-Rd. In addition, complete response rates and measurable residual disease (MRD) negativity were significantly higher with daratumumab-Rd. 116 Similarly, combining daratumumab with bortezomib and dexamethasone resulted in a significantly higher response rate, depth of response, and PFS in MM patients with at least one prior therapy, but not bortezomib-refractory disease. 117 Others investigated in a phase Ib study, the combination of daratumumab plus pomalidomide-dexamethasone in relapsed/refractory MM patients (89% lenalidomide-refractory, 71% bortezomib-refractory and 30% carfilzomib-refractory). ORRs were 60% with median PFS and OS of 8.8 months and 17.5 months respectively. 118 A recent phase III study describes the use of daratumumab in newly diagnosed, transplant ineligible patients, comparing bortezomib, melphalan and prednisone (VMP) with VMP plus daratumumab. VMP combined with daratumumab significantly increased ORR rates, complete response (CR) rates and improved PFS. 119 Daratumumab is currently approved by European Medicines Agency (EMA) and United States Food and Drug Administration (US FDA) as monotherapy in relapsed/refractory myeloma, and in combination with Rd and bortezomib-dexamethasone (VD) in patients with at least one prior line of therapy. The US FDA also approved daratumumab in combination with pomalidomide-dexamethasone (PD) for the treatment of MM patients who have received at least two prior therapies including lenalidomide and a proteasome inhibitor. In addition, the US FDA and recently the EMA, have approved daratumumab plus VMP for newly diagnosed patients who are ineligible for auto-SCT.
In addition to CD38 and SLAMF7, BCMA is an interesting target for MM therapy. BCMA is a member of the tumor necrosis factor superfamily, only expressed by some B-cells, normal plasma cells, and highly expressed on MM cells. Preclinical studies have shown that an anti-BCMA antibody conjugated to the microtubule disrupting agent monomethyl auristatin-F (antibody–drug conjugate named: GSK2857916) specifically inhibits MM cell activity and also induces effective ADCC against allogeneic and autologous MM cells. 120 This has led to a clinical phase II study (currently ongoing), which investigates this compound in heavily pretreated, relapsed/refractory MM patients. Preliminary results after a median of 6.6 months show an ORR of 60% and a median PFS of 7.9 months. 121
Chimeric antigen receptor T-cells
In a chimeric antigen receptor (CAR), the antigen-recognition part of a monoclonal antibody is combined with T-cell receptor domains and costimulatory domains. The CAR gene can be introduced in T-cells using viral transduction. This generates cells that can directly recognize (tumor) antigens, followed by an activation of the cytotoxic machinery of the T-cell causing kill of the target cells. 122 The advantages of CAR T-cells are the HLA-independent mode of action (as compared with unmodified T-cells), and a much more efficient kill compared with antibodies (ADCC). The expansion and activation of the CAR T-cells in the patients may lead to the so called cytokine-release syndrome. This is a potentially fatal clinical syndrome caused by a striking release of pro-inflammatory cytokines, and is characterized by fever, dyspnea, and hypotension and can lead to shock and multi-organ failure. However, the use of the anti-IL-6 antibody tocilizumab and corticosteroids is effective in the majority of patients.
Successful clinical results have been obtained using CD19 targeting CAR T-cells in B-cell malignancies.123,124 These results have led to the development of CAR T-cells for other targets.
In MM, the use of anti-BCMA CAR T-cells is currently being investigated in several studies (Table 1).125–131 In these studies, heavily pretreated, relapsed/refractory MM patients were treated with different doses of anti-BCMA CAR T-cells after a short course of leukocyte-depleting chemotherapy. High response rates were observed [⩾ partial response (PR) ranging from 77–100%], including some patients achieving a stringent CR. Cytokine-release syndrome occurred frequently (all grades ranging from 50–83%), but was well manageable.125–131 Another study investigated CAR T-cells targeting the kappa light chain in seven MM patients. 132 No objective responses were observed, but treatment with the CAR T-cells lead to stable disease ranging from 2–17 months, with no significant toxicities. 132 Several preclinical CAR T studies have shown promising results with other MM antigens, including CD38, CD44v6 and SLAMF7, forming the rationale for new clinical trials.81,133–135
Characteristics of trials evaluating BCMA-targeted CAR T-cells in MM.

Ag, antigen; BCMA, B-cell maturation antigen; CAR, chimeric antigen receptor; CR, complete response; CRS, cytokine-release syndrome; Cy, cyclophosphamide; EGFRt, truncated Endothelial Growth Factor Receptor; Flu, fludarabine; NA, not available; NCI, National Cancer Institute; NCT, ClinicalTrials.gov identifier; PR, partial response; scFv, single-chain variable fragment; sCR, stringent complete response.
Bispecific antibodies
Despite the promising clinical results with CAR T-cells, particularly CD19 targeting CAR T-cells in B-cell malignancies, their production requires the use of autologous cells and is considerably time-consuming. Bispecific antibodies (BsAbs) contain two antigen-recognition domains. One of them designed to recognize (e.g.) CD3, which is expressed on almost all T-cells, and the other targeting a tumor antigen. This technique enables bringing T-cells in proximity to tumor cells, causing T-cell proliferation and tumor cell lysis. BsAbs can be generated in large quantities and stored until use, creating an ‘off the shelf’ product. Bispecific T-cell engagers (BiTEs) are a type of BsAbs consisting of two single-chain variable fragments of different antibodies. A potential advantage of BiTEs, because of their shorter half-life compared with larger BsAb constructs, is the management of toxic side effects simply by halting the infusion. 136 In line with CAR T-cell studies, successful clinical results have been obtained with the CD3/CD19 BiTE blinatumomab targeting CD19 positive ALL,137,138 relapsed/refractory diffuse large B-cell lymphoma (DLBCL), and other types of NHL.139,140 In preclinical MM studies, several antigens were investigated as therapeutic target for BsAbs, including BCMA, CD38, CD138, and Fc receptor like 5 (Fcrl5).141,142 Based on promising preclinical results, several clinical trials with BiTEs and other BsAbs are ongoing in MM. The mode of action of BsAbs requires well-functioning autologous T-cells. However, MM is characterized by a defective immune system, potentially limiting their effectiveness. Furthermore, regulatory T-cells also express CD3 and can be activated upon the use of BsAbs further decreasing anti-tumor immune responses. 143
Immune-checkpoint inhibition
The development of antibodies blocking co-inhibitory receptors (checkpoint inhibitors) caused a major shift forward in cancer immunotherapy. Successful results using checkpoint inhibitors in relapsed/refractory Hodgkin’s lymphoma,144,145 led to studies in other hematological malignancies including MM. An increased expression of PD-L1 on malignant plasma cells has been observed in MM patients, together with elevated PD-1 expression on T-cells and NK cells.35,40,146,147 Despite these findings, monotherapy with the PD-1 inhibitor nivolumab had no clinical activity. 148 Nevertheless, in a phase I study the combination of pembrolizumab (an anti-PD-1 antibody), lenalidomide and dexamethasone had an ORR of 50% in heavily pretreated, relapsed/refractory MM patients, 149 with and ORR increasing to 60% when lenalidomide was replaced by pomalidomide in a phase II study. 150 However, at present all studies investigating the combination of IMiDs and PD-1/PD-L1 inhibitors are put on clinical hold due to an increased number of deaths in two phase III studies investigating the combination of pembrolizumab with pomalidomide in relapsed/refractory (KEYNOTE 183 study) and with lenalidomide in newly diagnosed MM (KEYNOTE 185 study). However, based on preclinical studies showing synergy between PD-1/PD-L1 inhibitors and antibodies targeting MM cell surface antigens, clinical trials evaluating PD1/PD-L1 inhibitors with other anti-MM agents, such as daratumumab, are ongoing (ClinicalTrials.gov identifiers: NCT01592370 and NCT03184194). 151
Dendritic cell vaccination
The ultimate goal of DC vaccination strategies is the in vivo induction or stimulation of tumor specific T-cell immunity against cancer cells in an antigen-dependent fashion, so that patients not only experience an anti-tumor effect, but also develop a long-term protection against a possible relapse of the disease. DCs are professional APCs capable of efficiently stimulating naïve T-cells to build up an anti-tumor response. 152 DC-based immunotherapy has been studied in a wide range of malignancies, and has been shown to be safe. 153 In MM, different strategies have been studied, including autologous DCs pulsed with idiotype, autologous DCs fused with MM cells, and autologous DCs loaded with myeloma-associated antigen mRNA (MAGE3, BCMA and survivin).154–157 Although the use of idiotype pulsed DCs and DCs loaded with myeloma-associated antigen mRNA resulted in the induction of antigen specific T-cells in a subset of patients, clinical responses were disappointing. However, promising results have been observed using DC-MM fusion vaccines. In a phase II clinical study, the use of DC-MM fusion vaccines after auto-SCT was shown to induce anti-MM CD4+ and CD8+ T-cells, with a conversion of a PR to CR following vaccination in 24% of patients. 154 This has led to the currently ongoing three-arm multicenter phase II randomized clinical trial, randomizing patients after upfront auto-SCT to (1) vaccination with a DC/MM cell fusion vaccine plus GM-CSF plus lenalidomide maintenance, (2) lenalidomide maintenance with GM-CSF, or (3) lenalidomide maintenance only (ClinicalTrials.gov identifier: NCT02728102). DCs are also of interest following allo-SCT, as it has been shown that the GVT effect depends on the presence of host DCs (capable of presenting host antigens).158–160 However, allo-SCT results in rapid replacement of host DCs by donor DCs, which may hamper the alloreactivity of donor lymphocytes. 161 Therefore, the GVT effect can theoretically be boosted by administration of DCs capable of presenting host antigens. We have published two phase I/II trials investigating the combination of donor lymphocyte infusions combined with DCs loaded with mHag after allo-SCT.162,163 In both trials, we were able to show the induction of mHag-specific T-cells after vaccination. However, clinical responses were disappointing, indicating the need for improvement of the current vaccination strategy.
Future perspectives
It is clear that immunotherapy has found its way to MM, adding significant improvement in survival of patients. Challenges that lie ahead are first of all the timing of these immunotherapeutic strategies in the disease course. While most of the studies were performed in the relapsed/refractory setting, it is likely that patients benefit the most when immunotherapy is applied at an earlier time point. Indeed, daratumumab combined with VMP is now approved for newly diagnosed patients after the successful results of the ALCYONE study. 119 However, the application of immunotherapy earlier in the disease course should take into consideration the potential toxicities of some of these treatments (e.g. with CAR T-cells) in addition to the costs. Secondly, and in line with the optimal timing, is the optimal sequence of this increasing number of effective immune therapies, which is currently unknown. Thirdly, optimal combination treatments targeting both the myeloma and the myeloma-associated immunosuppression, should be sought to further potentiate these therapies. Despite these challenges, we have already come quite far.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
NvdD: research support from Celgene, Janssen Pharmaceuticals, Novartis, BMS, and AMGEN; advisory boards for Celgene, Janssen Pharmaceuticals, Novartis, BMS, AMGEN, Takeda, Bayer, and Servier.
HML: Research support and honoraria: Celgene and Janssen Pharmaceuticals.
The other authors declare no conflicts of interest regarding this work.