
Abstract
The therapeutic options available for patients with multiple myeloma have greatly expanded over the past decade and incorporating these novel agents into routine clinical practice has significantly improved outcomes. The next generation of therapeutics is available for relapsed and refractory patients either as standard of care or in clinical trial, and these drugs represent a generational paradigm shift. Patients now have access to a multitude of novel immunotherapeutics, including monoclonal antibodies, an antibody–drug conjugate, chimeric antigen receptor T-cells (CAR-T), and bispecific T-cell redirecting antibodies, and novel oral therapies including selinexor (selective inhibitor of nuclear export) and venetoclax (bcl-2 inhibitor). While these drugs have the potential to be highly efficacious in certain subsets of patients when used as single agents or in combination regimens, they are each associated with unique toxicity profiles. It is imperative to understand these potential adverse events to ensure patient safety. Appropriate supportive care management is paramount to maximize drug exposure and therapeutic efficacy. The following review focuses its discussion on drugs and combination regimens that are currently FDA-approved and those that continue to be investigated in clinical trials, highlights the clinically relevant toxicity profiles for each of the different agents, and provides practical considerations for the treatment team.
Keywords
Introduction
Multiple myeloma (MM) is a hematologic cancer characterized by malignant transformation of plasma cells, a terminally differentiated B-cell. In 2021, there was an estimated ~34,000 new cases and ⩾12,000 deaths in the United States. 1 In most, MM is associated with production of a monoclonal protein and end-organ damage that may include renal dysfunction, anemia, bone lytic lesions, and hypercalcemia. Despite dramatic improvements in treatment options, the disease remains incurable, but patients treated with contemporary approaches have an improved overall survival over 10 years. 2
Foundational shifts in the MM treatment landscape have occurred over the last 20 years. The clinical development and FDA approval of immunomodulatory drugs (IMiDs) and proteosome inhibitors (PIs) in the early 2000s, along with the recent emergence of monoclonal antibodies (mAbs), were fundamental breakthroughs responsible for improving survival outcomes (Table 1). Other immunotherapeutics are now changing our approach to treatment. Antibody–drug conjugates (ADC), chimeric antigen receptor T-cells (CAR-T) and T-cell redirecting bispecific antibodies, alongside drugs with novel antitumor mechanisms, such as selinexor and venetoclax, are leading the next revolution and shifting us away from conventional chemotherapy toward effective, and in some cases, better tolerated interventions. However, these next-generation therapeutics are not without toxicities. Each drug class is associated with unique side effect profiles. Understanding these toxicities and initiating appropriate supportive and preventive strategies is critical for maximum drug exposure, improved response, and quality of life for our patients. In the present review, we highlight specific drugs within each of these novel drug classes to provide an overview of relevant side effect profiles and discuss practical management strategies.
Selected FDA approved therapies for the treatment of newly diagnosed and relapsed/refractory multiple myeloma from 2015-2022.

IMiD, immunomodulatory drugs; IV, intravenous; PI, proteosome inhibitor; SC, subcutaneous; RRMM, relapsed and refractory multiple myeloma; NDMM, newly diagnosed multiple myeloma; R, revlimid (lenalidomide); d, dexamethasone; V, velcade (bortezomib); P, pomalidomide; T, thalidomide; K, kyprolis (carfilzomib); aCD38 mAB, anti-CD38 monoclonal antibody.
FDA-approved next-generation therapeutics
Monoclonal antibodies
CD38 -directed mAbs: daratumumab (Darzalex® and Darzalex Faspro®) and isatuximab (Sarclisa®)
Daratumumab (human) and isatuximab (chimeric) are IgG1κ mAb targeting CD38.3,4 Single-agent intravenous (IV) daratumumab was FDA-approved in November 2015 and subcutaneous (SC) and IV daratumumab are approved in triplet and quad-regimens for the treatment of patients with newly diagnosed (NDMM) and relapsed and refractory (RRMM) multiple myeloma. Isatuximab was approved for the treatment of RRMM in combination with pomalidomide and dexamethasone, and carfilzomib and dexamethasone in 2020 and 2021, respectively (Table 1). While both are available in IV formulations, SC daratumumab and hyaluronidase human-fihj (Darzalex Faspro®) was approved in 2020 based on the COLUMBA trial that demonstrated the non-inferiority of the SC formulation.5–7 Isatuximab SC is currently being investigated (NCT04045795). 8 In practice, prescribers can use both forms of daratumumab interchangeably, but SC is generally preferred based on more convenient administration, including decreased time of drug administration, first-dose adverse reactions and post-first-dose monitoring requirements, and overall improved patient satisfaction.9–12 A selection of relevant clinical trials investigating daratumumab and isatuximab as single agents and in combination regimens highlights the most common any grade and grade ⩾ 3 adverse events associated with aCD38 mAb treatment (Table 2).13–22
Selected trials investigating monoclonal antibodies and most relevant toxicities.

ALT, alanine aminotransferase; AST, aspartate transaminase; IMiD, immunomodulatory drugs; IRR, Infusion-related reactions; PI, proteosome inhibitor; URI, upper respiratory tract infections; infections; HTN, hypertension; NDMM, newly diagnosed multiple myeloma, SOC, standard of care; Len, lenalidomide.
Daratumumab is infused intraveneously at a dose of 16 mg/kg and injected over 3–5 min at a fixed dose of 1800 mg weekly for two cycles, every other week for four cycles, and then monthly thereafter. In contrast, isatuximab is infused at a dose of 10 mg/kg weekly for cycle 1, followed by every other week thereafter. The infusion time for daratumumab is based on a progressive titration starting with 6.5 h at first dose followed by 4.5 h at second dose and 3.5 h thereafter, whereas isatuximab is slowly titrated up for the first two weekly injections and typically lasts 3–4 h barring any reactions or complications. To decrease infusion times, numerous trials have confirmed that rapid infusion of daratumumab 90 min and isatuximab 70 min can be initiated safely following evidence of tolerability during the first cycle (Table 3).23–29
Practical considerations associated with monoclonal antibody treatment.

IV, intravenous; IVIG, intravenous immunoglobulins; EPd, Elo + pomalidomide; ERd, Elo + lenalidomide; IRR, infusion-related reactions; SC, subcutaneous; ALC, absolute lymphocyte count; ANC, absolute neutrophil count; Dd, daratumumab + dexamethasone; DRd, daratumumab + lenalidomide + dexamethasone; DPd, daratumumab + pomalidomide + dexamethasone; IsaPd, isatuximab + pomalidomide + dexamethasone; IsaKd, isatuximab + carfilzomib + dexamethasone; G-CSF, granulocyte colony stimulating factor; DS, double strength; LLN, lower limit of normal; IgG, Immunoglobulin G; PO, by mouth; TMP-SMX, trimethoprimsulfamethoxazole.
Infusion-related reactions (IRRs) are common in all formulations but more likely with IV administration and occur primarily with the first dose (Table 3). Associated symptoms are generally low grade and include chills, fever, nausea, nasal congestion, cough, and dyspnea. The time to IRR onset is generally longer with SC versus IV daratumumab (3.4 versus 1.5 h, respectively), though low-grade, non-serious delayed IRR (incidence of IRR occurring the day after infusion/injection) have been reported for both daratumumab formulations but are rare.5,6 In general, those patients without IRR following IV daratumumab or isatuximab will not require post-dose monitoring, but if reactions are evident, we generally monitor patients for an additional 2 h after the completion of infusion. Patients treated with SC daratumumab are currently monitored for up to 4 h after the first dose and 1–2 h after the second dose though a recent retrospective study suggests that standard 30-min monitoring post-injection may be sufficient if adequate IRR prophylaxis is provided. 10
To mitigate the risk of IRR, split dosing may be considered with use of IV daratumumab, 30 however, utilization of an appropriate IRR prophylactic regimen prior to treatment with IV/SC daratumumab and isatuximab is essential to ensure safe drug administration. We recommend acetaminophen, diphenhydramine, dexamethasone, montelukast, and famotidine 15–60 min prior to drug administration (Table 3).31–33 After the initial cycle of either daratumumab formulation, famotidine and montelukast may be discontinued and a rapid taper of dexamethasone can be considered, especially in those patients that did not experience IRR, are steroid intolerant, or are receiving SC formulation.24,34,35 In addition, we do not recommend > 20 mg weekly dexamethasone or equivalent at any time for patients > 75 years of age. If IV daratumumab is preferred per patient preference or is used to reduce the minimal risk of delayed IRR associated with SC daratumumab, oral corticosteroids the day following infusion may be considered during the first cycle. In those patients treated with isatuximab, we utilize a similar prophylactic approach, though montelukast is not included and use of an H2 blocker is continued over the course of treatment. 29
Other than IRR, patients often experience hematologic toxicities, fatigue, dyspnea, upper respiratory tract infections (URI), including pneumonia, and gastrointestinal events, including diarrhea and constipation; the incidence and severity are ultimately dependent on the combination regimen used (Table 2). 36 Used as single agents, the most common all-grade treatment emergent adverse events (TEAEs) were URIs and arthralgias the addition of PIs are associated with higher incidence of peripheral neuropathy and thrombocytopenia, while the addition of IMiDs lead to more diarrhea, anemia, and neutropenia.13,15,18,19,21,22,37–40 In those patients treated with single-agent daratumumab or isatuximab, cytopenias (neutropenia, lymphopenia, anemia, and thrombocytopenia) were the most common grade 3–4 TEAEs, occurring primarily within the initial months of treatment initiation, and as expected, treatment with multi-agent regimens increased the incidence of these high-grade hematologic events. Notably, a prespecified subgroup analysis of COLUMBA indicated a higher incidence of neutropenia in patients with low body mass index (⩽ 65 kg) treated with SC daratumumab, however, this did not lead to a clinically notable increase in infections.5,41
Given the high incidence of associated neutropenia, lymphopenia, and hypogammaglobulinemia, patients are highly susceptible to bacterial and viral infections, most notably URI and pneumonia (Table 2). 36 Though data are limited, median time to first infection in a small case series of daratumumab treated patients was 2.5 months; range, 0.1–18.7 months. 42 Atypical infections are uncommon, however, reactivation of herpes zoster and Epstein–Barr virus/cytomegalovirus, pneumocystis jirovecii pneumonia (PJP), progressive multifocal leukoencephalopathy, bronchopulmonary aspergillosis, fungal meningitis, listeriosis, and disseminated cryptococcosis are reported.43–50 As expected, infections occur at a higher rate in those patients with higher grade neutropenia and lymphopenia, notably heavily pretreated patients treated with combination regimens within the initial 6 months of treatment. In this population treated with daratumumab, the median time to severe infection was ~50 days in patients with severe lymphopenia versus ~90 days in those without severe lymphopenia. 51
Given the increased risk of infection in these patients, all patients should receive antiviral prophylaxis with acyclovir or valacyclovir, PJP prophylaxis in the setting of prior PJP infection, heavily pretreated disease, or if the absolute lymphocyte count is below the lower limit of normal, and should be considered for intravenous immunoglobulins (IVIGs) in the setting of recurrent infections and IgG < 400 as this intervention has been associated with a 72% reduction in grade 3–4 infections. 42
Another important infectious consideration is the potential for hepatitis B virus (HBV) reactivation related to drug-associated natural killer (NK) cell depletion and suppression of humoral immunity.52–54 We recommend all patients be tested for HBV prior to starting mAb treatment and to undergo serial monitoring of liver enzymes over the course of treatment. Patients may continue mAb treatment alongside prophylaxis or preemptive therapy with either tenofovir or entecavir (not lamivudine due to low-resistance thresholds with this agent). 55 Monitoring and treatment of active hepatitis B disease requires consultation with a hepatologist to determine duration of therapy, appropriate frequency for trending viral load with PCR throughout treatment, and plan for maintaining prophylaxis against HBV once mAb therapy is completed. Patients with significant risk factors should also be screened for hepatitis C and HIV as clinically indicated. 56
Patients with MM are inherently at greater risk for infection and appropriate prevention strategies during the COVID-19 pandemic continue to be center stage for patients, family members, and care teams given this populations increased risk of adverse outcomes, including death.57–59 There is little doubt that the best protection from COVID-19 infection is prevention. Mask-wearing, hand-washing, avoidance of crowds, and sick individuals, and vaccination, even in patients potentially unable to mount an optimal vaccine response, are essential precautionary measures in all patients. We strictly follow the Centers for Disease Control guidelines for vaccination and encourage all of our patients to receive a full vaccination series, including the second booster. In addition, all patients are offered tixagevimab and cilgavimab (Evusheld™), a recombinant human IgG1κ mAb product that binds to non-overlapping epitopes of the spike protein receptor-binding domain of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and blocks attachment to the human ACE2 receptor, for pre-exposure prophylaxis of COVID-19. 60
SLAMF7-directed antibody: elotuzumab (Empliciti®)
Elotuzumab targets signaling lymphocytic activation molecule F7 (SLAMF7) or CS-1, a glycoprotein expressed on the surface of more than 95% of bone marrow myeloma and NK cells. 61 Elotuzumab is an IV infusion first approved in 2015 in combination with lenalidomide and dexamethasone based on the ELOQUENT-2 trial for patients previously treated with one to three prior lines of therapy,17,38,39 and in 2018, was approved in combination with pomalidomide and dexamethasone (EPd) based on the ELOQUENT-3 trial.16,62 Treatment with both triplet regimens is based on 28-day cycles and elotuzumab is infused weekly for two cycles. Starting in cycle 3, when combined with lenalidomide, elotuzumab is infused every other week but in combination with pomalidomide, it is only infused on the first day of each cycle (Table 2).
Elotuzumab is associated with a much lower rate of IRR than the IV formulations of daratumumab and isatuximab. Data from the ELOQUENT-2 and 3 trials demonstrated a 5–11% rate of all-grade IRR, events were all grade 1 or 2 except for five patients that had a grade 3 event on ELOQUENT-2.16,39 Similar to daratumumab and isatuximab, the majority of these events were associated with fever, chills, cough, congestion, and nausea. 38 Similar to the other mAb, pretreatment with acetaminophen, diphenhydramine, famotidine, and dexamethasone is indicated 15–60 min prior to infusion, though different from the recommended dosing per label, we utilize standard dexamethasone dosing of either 20 or 40 mg based on age. 32 Other TEAE related to elotuzumab are similar to the anti-CD38 mAb and vary depending on the combination treatment regimen. For example, the most common grade 3 or higher events of triplet therapy included lymphopenia (77%), neutropenia (34%), infections (33%), anemia (20%), pneumonia (14%), fatigue (10%), and diarrhea (8%) on ELOQUENT-2, and infections (13%), neutropenia (13%), anemia (10%), thrombocytopenia (7%), and pneumonia (5%) on ELOQUENT-3 (Table 2). Our approach to IRR and infection prophylaxis associated with elotuzumab is similar to that described for the anti-CD38 mAb (Table 3).
B-cell maturation antigen-directed novel immunotherapeutics
ADC: belantamab mafodotin (BLENREP®)
Belantamab mafodotin is a first-in-class immunoconjugate or ADC consisting of an afucosylated humanized IgG1 B-cell maturation antigen (BCMA)-directed mAb conjugated to the microtubule-disrupting agent monomethyl auristatin F (MMAF). Belantamab mafodotin was FDA-approved in 2020 for patients with RRMM previously treated with ⩾ 4 prior therapies, including an IMiD, PI, and an anti-CD38 mAb based on the DREAMM2 trial, a randomized phase II trial investigating two different doses of belantamab in heavily pretreated patients median of seven prior lines of treatment.63,64 Based on the efficacy and safety profile, single-agent belantamab 2.5 mg/kg infused every 3 weeks was approved. Overall, it is considered a generally well-tolerated, steroid-sparing treatment, though it is associated with manageable corneal and hematologic toxicities.
DREAMM2 confirmed that microcyst-like epithelial changes (MECs) were common. 65 In those patients treated at the 2.5 mg/kg dose on DREAMM2, 72% of patients had any grade keratopathy (46% grade ⩾ 3) with or without symptoms or changes in best-corrected visual acuity (BCVA) that occurred on average 37 days (range, 19–143 days) after starting treatment. In addition, 54% had any grade (31%, grade ⩾ 3) BCVA changes, 18% had a meaningful decline in the Snellen Visual Acuity to 20/50 or worse that recovered within an average of 21.5 days, 25% had any grade blurred vision (4%, grade ⩾ 3), and 15% experienced any grade dry eyes (1%, grade ⩾ 3).63,65 In those patients with grade ⩾ 2 keratopathy, 77% recovered on average 86.5 days; range, 8–365 days after their initial ocular event. 65 No patient had permanent vision changes or loss, likely accounted for by the continuous regeneration of the corneal epithelial layer.64,65 Notably, in responding patients, 80% maintained their response even after dose holds over 63 days, 66 indicating that responses are durable even when drug dosing is modified.
Ultimately, clinical investigation of alternate dosing regimens will be instrumental in defining the best tolerated and efficacious treatment schedule, 67 but at this time, per requirements of the BLENREP REMS Program, treatment requires corneal examinations by an ophthalmologist or optometrist at baseline and before each treatment that include, at a minimum, assessment of BCVA and keratopathy by slit lamp examination. Use of preservative free eye drops (available for free through the GlaxoSmithKline BLENREP Eye Drop Supportive Care Program and avoidance of contact lenses while on treatment are also important practical considerations (Table 4). 68
Belantamab mafodotin practical considerations.

Other noteworthy belantamab-associated all-grade toxicities include cytopenias (thrombocytopenia (38%), anemia (27%), and lymphopenia (14%)), pyrexia (23%), fatigue (16%), and aspartate transaminase (AST) increase (21%), and the most common grade ⩾ 3 events include keratopathy, 46%; thrombocytopenia, 22%; anemia, 21%; lymphopenia, 13%; and neutropenia, 11%. 63 Overall, the toxicities associated with belantamab treatment are manageable, though ocular toxicities can be profound and special attention to management of ocular adverse events is critical for safe drug administration and further development of this agent in multi-drug combinations (Table 5).
Belantamab dose modifications.

BCVA, best-corrected visual acuity; AE, adverse event
CAR-T: chimeric antigen receptor T-cells; idecabtagene vicleucel (Abecma®, Ide-cel) and ciltacaptagene autoleucel (Carvykti®, Cilta-cel)
The clinical development of multiple b-cell maturation antigen (BMCA)-directed CAR-T constructs, namely, Ide-cel and Cilta-cel, highlight two of the many novel T-cell adoptive therapies available commercially and in clinical trial. Ide-cel was FDA-approved in 2021, based on the KARMMA platform, for patients previously treated with at least four prior lines of therapy, including an IMiD, PI, and anti-CD38 mAb. 69 Cilta-cel, developed on the CARTITUDE platform, is associated with impressive responses, 70 and was recently FDA-approved in early 2022 (Table 6). The toxicities associated with CAR-T in RRMM reflect the CAR-T experience in other hematologic malignancies and are characterized by cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). In addition, cytopenias, including B-cell aplasia, hypogammaglobulinemia associated with the need for infection prophylaxis, and hemophagocytic lymphohistiocytosis (HLH) are major considerations in routine practice (Table 7).
Selected next generation clinical trials investigating novel immunotherapeutics.

Denotes number of patients treated at the recommended phase 2 dose (RP2D) and associated toxicity profile. ALT, alanine aminotransferase; AST, aspartate transaminase; BCMA, B-cell maturation antigen; CAR-T, chimeric antigen receptor T; CRS, cytokine release syndrome; mTTO, median time to onset; NR, not reported; NT, neurotoxicity.
Practical considerations associated with CAR-T and bispecific T-cell engager.
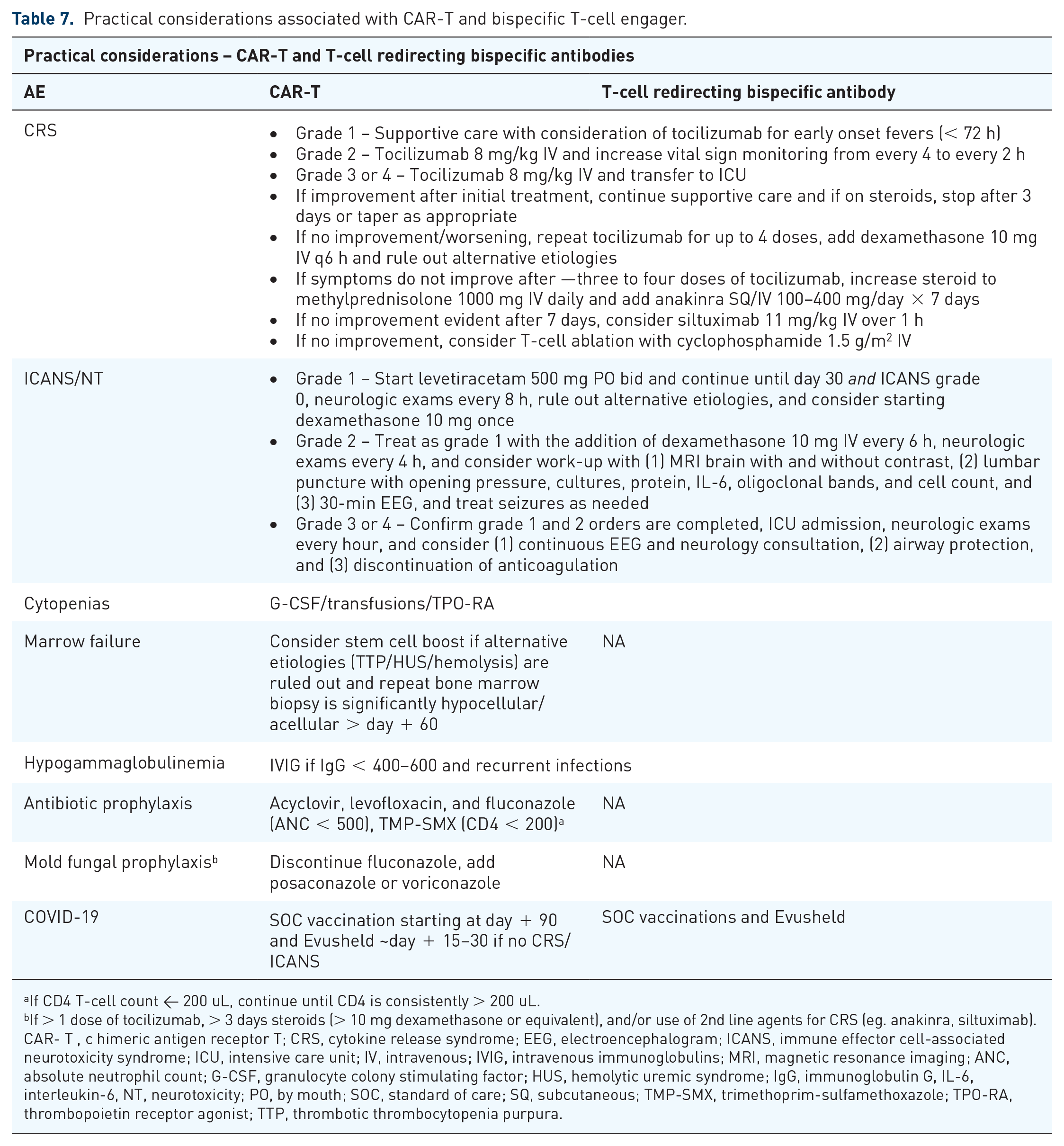
aIf CD4 T-cell count < 200 uL, continue until CD4 is consistently > 200 uL. bIf > 1 dose of tocilizumab, > 3 days steroids (> 10 mg dexamethasone or equivalent), and/or use of 2nd line agents for CRS (eg. anakinra, siltuximab). CAR- T , c himeric antigen receptor T; CRS, cytokine release syndrome; EEG, electroencephalogram; ICANS, immune effector cell-associated neurotoxicity syndrome; ICU, intensive care unit; IV, intravenous; IVIG, intravenous immunoglobulins; MRI, magnetic resonance imaging; ANC, absolute neutrophil count; G-CSF, granulocyte colony stimulating factor; HUS, hemolytic uremic syndrome; IgG, immunoglobulin G, IL-6, interleukin-6, NT, neurotoxicity; PO, by mouth; SOC, standard of care; SQ, subcutaneous; TMP-SMX, trimethoprim-sulfamethoxazole; TPO-RA, thrombopoietin receptor agonist; TTP, thrombotic thrombocytopenia purpura.
CRS is a clinical syndrome resulting from induction of inflammatory cytokines associated with T-cell activation and proliferation. It is most commonly associated with relatively low-grade clinical manifestations, including fever, rigors, tachycardia, shortness of breath requiring oxygen, and hypotension. While the majority of patients experience low-grade toxicities per the Lee et al. 71 criteria, life-threatening symptoms with multi-organ involvement can occur. Based on the KARMMA experience, 84% of patients developed any grade CRS, 95% were grade 2 or less, and 52% received tocilizumab, a potent mAb inhibitor of the IL-6 receptor. The median time to CRS onset was 1 day; range, 1–12 days and symptoms lasted a median of 5 days; range, 1–63 days. 69 These findings are consistent with the CARTITUDE experience, in that 95% of patients treated with Cilta-cel experienced any grade CRS associated with 96% that had grade 2 or less events. Interestingly, the median time to CRS onset in these patients was 7 days (range, 5–8 days), median duration of CRS was 4 days (range, 3–6 days), and almost 70% of patients were treated with tocilizumab (Table 3). While tocilizumab is the most commonly used intervention for CRS, corticosteroids (usually dexamethasone) and anakinra, an IL-1 receptor antagonist, may also be utilized given their ability to suppress the inflammatory cascade.
HLH, also known as macrophage-activation syndrome (MAS), is another complication of CAR-T therapy and was present in 4–8% of patients in the Ide-cel dataset. 69 This entity is difficult to distinguish from CRS as both present with high fevers, organ toxicity, cytopenias, hyperferritinemia, and high levels of inflammatory cytokines. Often, onset is just after development of or in conjunction with CRS. Diagnosis is made with criteria described by the CARTOX working group, which is based on high ferritin levels > 10,000 and lack of response to treatment instituted for CRS. 72 In cases not responding to tocilizumab after 48 h, consideration is given for treatment of presumed HLH with anakinra or etoposide while further work-up is completed.
ICANS is the second most common immune cell-mediated toxicity associated with CAR-T. Clinical manifestations may include headache, aphasia, tremors or seizures, encephalopathy, and death resulting from cerebral edema. The pathology underlying ICANS/neurotoxicity is not well understood but may be related to cytokine crossover to the cerebrospinal space through disruption of the blood–brain barrier, allowing for immune cell trafficking into the central nervous system and further inflammatory cascade activation.73,74 In KARMMA, 18% of patients were reported to have all-grade ICANS/neurotoxicity of which 97% were ⩽ grade 2 and occurred at a median of 2 days (range, 1–10). 69 In CARTITUDE-1, 17% of patients were reported to have any grade ICANS with two patients experiencing a grade 3 or higher event. This occurred at a median of 8 days; range, 6–8 days. Perhaps uniquely, five patients reportedly developed movement disorders, neurocognitive events, and personality changes MNT after recovery from CRS and/or typical ICANS. 75 Common features of this group included high tumor burden, grade ⩾ 2 CRS, ICANS, and high CAR-T cell expansion/persistence. Lab features included higher absolute lymphocyte counts, absolute CD4+ T-cells and CAR-T cell persistence at days 14, 21, and 28. Higher peak levels of IL-6 and interferon gamma (IFN-γ) were also seen in this group. Treatment for ICANS includes first-line therapy with corticosteroids and then tocilizumab, siltuximab, or anakinra. Those receiving Cilta-cel and who develop MNT-associated symptoms may be less responsive to steroids, however, hence early stratification of these patients to receive other cytokine therapy beyond steroids/tocilizumab, appropriate full neurologic and infectious evaluation, including CSF exam and seizure prophylaxis are recommended. 75
The risk of prolonged cytopenias, including B-cell aplasia, is not uncommon. Grade 3–4 cytopenias lasting greater than 30 days after CAR-T infusion has been reported in 20–40% of patients in patients with non-Hodgkin lymphoma (NHL) and acute lymphoblastic leukemia (ALL).76–80 In regard to Ide-cel, prolonged neutropenia was seen in 34–49% of patients depending on T-cell dose with 85% of patients taking a median of 1.9 months to recover counts. Prolonged thrombocytopenia occurred in almost half of the patients with a median duration of 2.1 months. There is currently no consensus on the routine use of growth factors and thrombopoietin receptor agonists, but both may be considered, especially in those with infections or bleeding tendencies. Consideration should be given to the separation of G-CSF from CAR-T products by at least 3 weeks or until resolution of CRS. If these interventions fail, CD34 + stem cell boosts may be a viable option for rescue after bone marrow and peripheral blood evaluation has ruled out HLH and other causes of cytopenias including thrombotic thrombocytopenic purpura/hemolytic uricemia syndrome.
Because of off-target effects resulting from B-cell aplasia, immunoglobulin levels may be suppressed in patients following CAR-T therapy and for this reason, patients remain at high risk for infections and associated complications. We recommend that family and caregivers receive influenza and COVID-19 vaccinations prior to infusion of lymphodepleting chemotherapy for this reason. In addition, antiviral and PJP prophylaxis are provided through 6 months or longer post-CAR-T infusion if the CD4 count remains < 200 cells/uL (checked monthly). The use of antibacterial and antifungals may be considered in cases of prolonged neutropenia. In those treated for CRS or ICANS with corticosteroids, tocilizumab, or other agents, we routinely add fungal prophylaxis for 30 days after the last dose. Immunoglobulin replacement therapy is also instituted routinely for our patients if their IgG level is below 400–600 mg/dl and checked on a monthly basis, especially if recurrent infections are present (Table 7).
Selective inhibitor of nuclear export (SINE): selinexor (Xpovio®)
Selinexor is an oral selective inhibitor of nuclear export blocking exportin 1 (XPO1), a nuclear exporter overexpressed in MM cells that shuttles tumor suppressor proteins, the glucocorticoid receptor, and oncoprotein messenger RNAs out of the nucleus.81,82 Selinexor was initially FDA-approved in 2019 for the treatment of patients previously treated with last least four prior lines of therapy, including lenalidomide, pomalidomide, bortezomib, carfilzomib, and an anti-CD38 mAb based on the STORM trial. 83 In 2020, based on the randomized phase III BOSTON trial, selinexor combined with bortezomib was approved for patients previously treated with at least one prior line of therapy 84 and a recent meta-analysis showed that selinexor combined with a PI not only improves response but also reduces the incidence of common all-grade toxicities. 85 Based on results from STOMP,86–89 the National Comprehensive Cancer Network (NCCN) guidelines now lists multiple selinexor-based combinations as ‘Useful in Certain Circumstances for Early Relapses (1–3 prior therapies)’, 56 supporting use of these regimens in routine clinical practice (Table 8).
Other selected next generation therapeutics.

URI, upper respiratory tract infection; infection; HTN, hypertension; NR, not reported; SOC, standard of care; TLS, tumor lysis syndrome.
Selinexor combination therapy is associated with gastrointestinal toxicities, including nausea, vomiting, and diarrhea, decreased appetite and weight loss, fatigue, cytopenias, and URI. 85 As expected, the incidence and severity of these events vary based on selinexor dosing and combination therapy, and in general, patient education and significant supportive care, especially over the initial two cycles are required to maximize tolerability and drug exposure. 90
Appropriate dosing is critical and use of selinexor has now shifted from twice weekly to once weekly dosing when used in triplet regimens. 90 When used in combination triplet regimens, we recommend initiating selinexor 60–80 mg weekly during the first cycle and escalating to 100 mg weekly if tolerated. Supportive measures include use of low-dose olanzapine (2.5–5.0 mg) nightly for 3 days starting the day of selinexor, use of 5-HT3 receptor antagonist (most commonly ondansetron), and consideration of neurokinin-1 receptor antagonist (rolapitant). Based on drug tolerability, these anti-emetics may be weaned off following two cycles of treatment. We recommend obtaining at least weekly complete blood counts with dose holds and modifications as necessary, and G-CSF, transfusion support, and consideration of thrombopoietin receptor agonists and erythropoietin stimulating agents when applicable. Patients should also complete a once weekly metabolic panel. We instruct patients to maintain adequate hydration with at least 2 liters of fluid daily, ideally inclusive of salt-containing drinks, though in patients unable to tolerate this level of fluid intake, we consider use of salt tabs. In addition, we encourage patients to utilize anti-diarrheals (loperamide or diphenoxylate/atropine, or both) as necessary, consume adequate calories, consider use of appetite stimulants, and have a low threshold for consultation with a nutritionist. Ultimately, time and experience have proven that early and efficient supportive care leads to improved tolerability and allows patients to stay on treatment, if responding (Table 9).
Selinexor practical considerations.

*Should be started in all patients before treatment and continued for 1-2 cycles; SC, subcutaneous; ANC, absolute neutrophil count; G-CSF, granulocyte colony stimulating factor; IVF, intravenous fluids; PO, by mouth; SOC, standard of care.
Next-generation non-FDA-approved therapeutics in clinical trials
T-cell redirecting bispecific antibodies
Current therapies aimed at overcoming the profound immune dysfunction associated with RRMM are proving highly effective even in heavily pretreated patients. T-cell redirecting bispecific antibodies are an off-the-shelf, steroid-sparing, novel immunotherapy drug class with great potential to change the MM treatment landscape. The constructs closest to routine clinical use are designed to simultaneously bind CD3 on T-cells and a specific target epitope on the MM cell the current majority target BCMA and are still investigational at this time, these include AMG701, 91 teclistamab, 92 elranatamab, 93 ABBV-383B, 94 and REGN5458, 95 while talquetamab targets GPRC5D 96 (Table 6). Despite the fact that there are currently no FDA-approved bispecific antibodies, these drugs are moving through phase I and II trials and are actively being investigated in randomized phase III trials.91,93,94,96–98
T-cell redirecting bispecific antibodies are associated with response rates in the 60–80% range even as single agents.91–98 As a drug class, they are generally well-tolerated and associated with a relatively consistent side effect profile, including significant all-grade cytopenias, nausea, diarrhea, fatigue, transient increases in aspartate transaminase (AST) and alanine aminotransferase (ALT) (Table 3), and risk of low-grade neurotoxicity. Low-grade CRS, however, is the hallmark of these drugs. All of these agents are associated with grade 1 and 2 events with a typical onset within 1–2 days of initiating treatment and generally recognized during ramp-up and with the initial full-strength dosing. The clinical presentation of these events is consistent with CAR-T therapy, however, intervention with tocilizumab with or without steroids is less frequently needed (Table 4). Current clinical investigations of the bispecifics aim to decrease CRS incidence using step-up dosing, shifting to SC injections rather than infusion, and utilization of early intervention with tocilizumab with or without steroids. If successful, these highly effective drugs will likely take over the early relapsed space and be investigated in newly diagnosed patients.
BCL-2 inhibitor: venetoclax (Venclexta®)
Venetoclax is an oral, small molecule, highly selective BH3 mimetic that induces apoptosis by displacing proapoptotic proteins from bcl-2 and is the first targeted therapy for the treatment of MM. 99 This bcl-2 inhibitor is not yet FDA-approved despite national guideline recommendations supporting its use in combination with dexamethasone for patients with t(11;14) disease. 56 Currently, clinical trials are investigating venetoclax in combination with bortezomib, carfilzomib, and daratumumab for patients with bcl-2-dependent disease harboring t(11;14) or high bcl-2 expression (Table 5).100–104
In general, patients treated with venetoclax commonly experience diarrhea, nausea, and cytopenias that are managed per standard of care measures. However, it is evident that single-agent and combination venetoclax-based regimens are associated with an increased risk of potentially life-threatening infections. The BELLINI trial was a randomized phase III trial that investigated venetoclax in combination with bortezomib and dexamethasone versus the bortezomib and dexamethasone. 104 Results from this trial indicated impressive responses, but the experimental arm was associated with an unexpectedly high mortality rate. Subanalysis of patients with t(11;14) disease showed improved responses, and the addition of venetoclax was not associated with increased rates of mortality in this population versus those treated with bortezomib and dexamethasone alone. Ultimately, the results of BELLINI halted the approval of venetoclax combination therapy though it substantiated its use in patients with t(11;14) disease. Based on these results and data derived from other trials with venetoclax-based combination regimens,101–103 high-grade pneumonia remains one of the most concerning TEAE. As such, the more recent M15-538 trial mandated antibiotic prophylaxis though rates of pneumonia did not differ from the venetoclax monotherapy experience. 93 Overall, appropriate patient selection and use of vigilant supportive care, most notably, early detection and treatment of pneumonia and gastrointestinal toxicities, may allow effective use of this novel therapy for patients with t(11;14) MM (Table 10).
Venetoclax practical considerations.

PJP, pneumocystis jirovecii pneumonia; TLS, tumorlysis syndrome; TMP-SMX, trimethoprim sulfamethoxazole.
Conclusion
Since the FDA approval of daratumumab in 2015, the therapeutic options in the RRMM pipeline have greatly expanded. Incorporating these novel agents into established treatment combinations portends deeper responses and longer survival in the relapsed and refractory setting. Fortunately, many of these agents are less toxic than drugs historically used in the late-line setting and tolerable side effect profiles are allowing more patients access to a wider range of therapeutic options over the course of their disease. Driving into the future, utilization of our ever-expanding MM treatment toolbox and toxicity management will become increasingly complex as we continue to explore our armamentarium of novel therapeutics. While this massive momentum carries great promise for transforming myeloma into a chronic and treatable condition in many patients, consideration of interventions to offset the growing list of unique toxicities is paramount to enhancing both the quality and longevity of our patient’s lives. While this review provides a framework for the present and near future, there is no doubt that toxicity management strategies will continue to take center stage, and great attention must be paid to ensure that we maximize the quality of life of those patients under our care.
Footnotes
Acknowledgements
The authors express their special thanks to Claire Davies, MS, Strategic Communications at the Huntsman Cancer Institute, for her generosity and willingness to assist with graphic design. They also greatly appreciate the work of Michael Filtz, PharmD and Catherine Lee, MD, as they defined their institutional CRS/ICANS algorithms and graciously allowed them to include their work in this review.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M.S. is on the speakers’ bureau for GlaxoSmithKline, Karyopharm Therapeutics, and Janssen Pharmaceuticals. K.J. and B.L.M. have no relevant disclosures. D.W.S. is an advisor/consultant for Janssen Pharmaceuticals, Celgene, Abbvie, Sanofi, and GlaxoSmithKline.
Availability of data and materials
Not applicable.