
Abstract
Background:
Patients with lower-risk myelodysplastic syndromes (MDS) may experience anemia and a high transfusion burden, alongside a risk of progression to acute myeloid leukemia. Luspatercept, a recombinant fusion protein that acts as an erythroid maturation agent, was FDA/EMA-approved in 2020 based on the phase III MEDALIST trial. There remains an unmet need for anemia treatment in Asian patients for whom red blood cell (RBC) transfusion is a standard of care, and in whom rates/severity of anemia and serum erythropoietin levels are often higher versus Western patients.
Objectives:
The objective of this study was to assess the efficacy, safety, and tolerability of luspatercept in Asian patients with anemia due to transfusion-dependent lower-risk MDS with ring sideroblasts.
Design:
This was a phase II, single-arm, interventional bridging study (NCT04477850).
Methods:
Patients from China and Japan with very low-, low-, or intermediate-risk MDS with ring sideroblasts who were RBC transfusion-dependent received subcutaneous luspatercept starting at 1.0 mg/kg every 3 weeks. The primary endpoint was RBC transfusion independence (TI) ⩾8 weeks (weeks 1–24).
Results:
There was a statistically significant, clinically meaningful improvement of anemia in Asian patients; 60% (n = 18, p < 0.0001) achieved RBC-TI for ⩾8 weeks and 43% (n = 13) for ⩾12 weeks (weeks 1–24). Safety was consistent with the known profile of luspatercept in MDS.
Conclusion:
These results support luspatercept as a well-tolerated, efficacious alternative to transfusions for Asian patients with lower-risk MDS, who tend to have more severe anemia.
Introduction
Myelodysplastic syndromes (MDS) are bone marrow neoplasms characterized by ineffective erythropoiesis; peripheral cytopenia, particularly anemia; and an increased risk of progression to acute myeloid leukemia.1–3 In Asia, the incidence of MDS is rising rapidly due to the aging population and improved diagnosis. The incidence of MDS in China is estimated at 1.5/100,000, 4 and in Japan, the incidence of MDS in 2008 was 3.8/100,000 for men and 2.4/100,000 for women. 5
At the conception of the International Prognostic Scoring System-Revised (IPSS-R) criteria, most patients with MDS were classified as lower risk (LR-MDS). 6 In LR-MDS, 85% of patients develop chronic anemia that has the biggest impact on morbidity and quality of life; therefore, treatment focuses on anemia management. With time, 40% of patients become dependent on red blood cell (RBC) transfusions, which bring additional risk.7,8 Long-term RBC transfusion dependence has several detrimental clinical effects including iron overload, cardiovascular symptoms, and complications,9–13 and a negative impact on survival. 14
Therapeutic goals of LR-MDS include the treatment of anemia, elimination or reduction of the need for RBC transfusions, improving health-related quality of life, and survival. However, therapeutic options for anemia treatment in patients with LR-MDS in regions of Asia, including China and Japan, are limited and include erythropoiesis stimulating agents (ESAs), such as epoetin (EPO) or darbepoetin (with or without hematopoietic growth factors like granulocyte colony-stimulating factor (G-CSF) or granulocyte-macrophage colony-stimulating factor), and administration of RBC transfusions.15,16 However, use of these agents can be further complicated by a lack of regulatory approval (e.g., ESAs in China), a lack of coverage by national health insurance (e.g., the addition of G-CSF to darbepoetin alpha in Japan), or limited availability (e.g., RBC transfusion supply in China). 15
Taken together, these represent an unmet need, and alternative treatments are a priority. One of them is luspatercept, a first-in-class recombinant fusion protein that binds select endogenous transforming growth factor β superfamily ligands and thus diminishes downstream Smad2/3 signaling. Luspatercept restores erythropoiesis by promoting erythroid expansion and maturation (acting at multiple stages of erythropoiesis, including early, mid, and late stages).17–19
MEDALIST is a completed phase III, randomized, double-blind, placebo-controlled multinational study (excluding Asian countries) to compare the efficacy and safety of luspatercept for the treatment of patients with anemia due to very low- to intermediate-risk MDS (by IPSS-R criteria) with ring sideroblasts (RS) who require RBC transfusions and who are refractory, intolerant, or ineligible to receive an ESA.20,21 Most patients achieving RBC transfusion independence (RBC-TI) for ⩾8 weeks through weeks 1–24 and/or modified hematologic improvement–erythroid (mHI-E) with luspatercept in the MEDALIST study had multiple responses with durable clinical benefit superior to placebo, including those with a high baseline transfusion burden. 20 The responses observed in MEDALIST were shown to be durable over extended follow-up. A median cumulative duration of response of RBC-TI ⩾8 weeks was 110.14 weeks, observed over a median follow-up period of 39.9 months with luspatercept; 59% of patients had a cumulative duration of RBC-TI ⩾8 weeks greater than a year. 22
Luspatercept was approved for use in patients with LR-MDS-RS after ESA failure by the US Food and Drug Administration and European Medicines Agency in 2020 based on the MEDALIST trial 21 and more recently was approved for use as a first-line therapy by the Food and Drug Administration and European Medicines Agency based on results from the COMMANDS trial. 23 However, there remains an unmet need in Asian patients who are unresponsive or refractory to ESAs (and patients in China, where ESAs are not approved for use in MDS) or for whom RBC transfusion is the sole standard of care. Toward this, luspatercept has recently been approved in Japan for patients with MDS-related anemia regardless of RS status or RBC transfusion dependency. 24 Additionally, rates and severity of anemia and serum EPO levels are often higher in Asian versus Western patients.25,26 However, it is unknown whether regional differences in baseline disease characteristics have an impact on the efficacy of luspatercept in Asian patients compared with Western patients.
Here, we report the results on a single-arm bridging study on the efficacy, pharmacokinetics (PK), and safety of luspatercept in Chinese and Japanese patients with anemia due to LR-MDS.
Methods
Study design and ethics
This phase II, multicenter, single-arm bridging study (NCT04477850) evaluated the efficacy, PK, and safety of luspatercept for the treatment of anemia due to very low-, low-, or intermediate-risk (by IPSS-R criteria) MDS with RS in Chinese and Japanese patients who require RBC transfusions.
The study enrolled patients at 14 sites in China and Japan and was approved by the institutional review board/independent ethics committee at each site. Patients had luspatercept administered subcutaneously every 3 weeks for at least 24 weeks. The starting dose of luspatercept was 1.0 mg/kg of body weight. The dose could be titrated stepwise to 1.33 mg/kg, then to 1.75 mg/kg (Supplemental Table 1). Best supportive care could be used in combination with study treatment when clinically indicated by the investigator.
Disease assessment occurred at week 25. Patients without clinical benefit discontinued luspatercept and entered follow-up. Patients with evidence of clinical benefit and absence of disease progression (per International Working Group (IWG) criteria for altering the natural history of MDS) could continue treatment in the extension phase until unacceptable toxicity, disease progression, withdrawal of consent, or other protocol-defined treatment discontinuation criteria are met.
A protocol amendment in September 2021 affected the study design, including clarifications to select inclusion criteria and the addition of exploratory endpoints in China patients only (reduction in RBC units transfused over 24 weeks compared with baseline; and mean hemoglobin (Hb) change over 24 weeks).
Patients
Eligible patients were 20 years of age or older and had a MDS with RS according to World Health Organization (WHO) 2017 criteria (i.e., with either ⩾15% RS or ⩾5% RS if an SF3B1 mutation was present and with <5% bone marrow blasts) 27 ; had IPSS-R—defined as very low, low, or intermediate-risk MDS without chromosome 5q deletion (del[5q]); had been receiving regular RBC transfusions (⩾2 units of packed RBCs per 8 weeks during the 16 weeks before luspatercept treatment); a Hb level of ⩽9.0 g/dL with anemia symptoms, or ⩽7 g/dL without anemia symptoms; and had no consecutive 56-day transfusion-free period 16 weeks before luspatercept treatment. Eligible patients were refractory or intolerant to prior ESA treatment (with documentation of nonresponse, response that was no longer maintained, or discontinuation of an ESA treatment given as single agent or as part of combination therapy at any time after introduction due to intolerance or an adverse event) or were ESA ineligible (based on a low chance of response to ESAs with serum EPO >200 U/L in patients not previously treated with ESAs). Additional eligibility criteria details are listed in the Supplemental Appendix.
Endpoints
The primary endpoint was RBC-TI for ⩾8 weeks, assessed during weeks 1–24. The secondary endpoints included RBC-TI for ⩾12 weeks, assessed during weeks 1–24, mHI-E per IWG criteria (2006; weeks 1–24); reduction in RBC units transfused over 16 weeks compared with baseline (weeks 9–24); mean Hb increase of ⩾1.0 g/dL (weeks 1–24); duration of RBC-TI ⩾8 weeks (weeks 1–24; week 1 to end of treatment (EOT)); mean decrease in mean serum ferritin over 16 weeks compared with baseline (weeks 9–24); time to RBC-TI for ⩾8 weeks (weeks 1–24); progression to acute myeloid leukemia; overall survival (OS); safety; PK; and antidrug antibodies. Key exploratory endpoints were reduction in RBC units transfused over 24 weeks compared with baseline (China patients only) and mean Hb change over 24 weeks (China patients only). Full definitions of endpoints are listed in Supplemental Table 2.
Statistical analysis
The sample size was based on the RBC-TI ⩾8-week response rate (weeks 1–24) and calculated using an exact test for single proportion (response rate ⩽13% vs >13%, based on pivotal MEDALIST study results), with a two-sided alpha of 0.05, and was determined to be 30. This provided 86% power to justify the RBC-TI ⩾8-week response rate (around 38%) of luspatercept higher than the estimated control of 13% (based on MEDALIST results). Patients who did not have transfusion independence for at least 8 weeks by week 24 were considered nonresponders for the primary endpoint. The percentages of patients with a primary endpoint response were compared with those obtained in MEDALIST using an exact test. The duration of transfusion independence of 8 weeks or longer was estimated using Kaplan–Meier analysis of the longest period of response at any dose.
All efficacy analyses were conducted in the intent-to-treat population (ITT; all enrolled patients). Safety analyses were conducted in enrolled patients who received at least one dose of luspatercept (safety population). PK analyses were conducted in enrolled patients who received at least one dose of luspatercept and had at least one measurable concentration of luspatercept. An exploratory analysis of the primary endpoint and main secondary endpoints in Chinese and Japanese patients was conducted, and results were reported descriptively.
Results
Patients
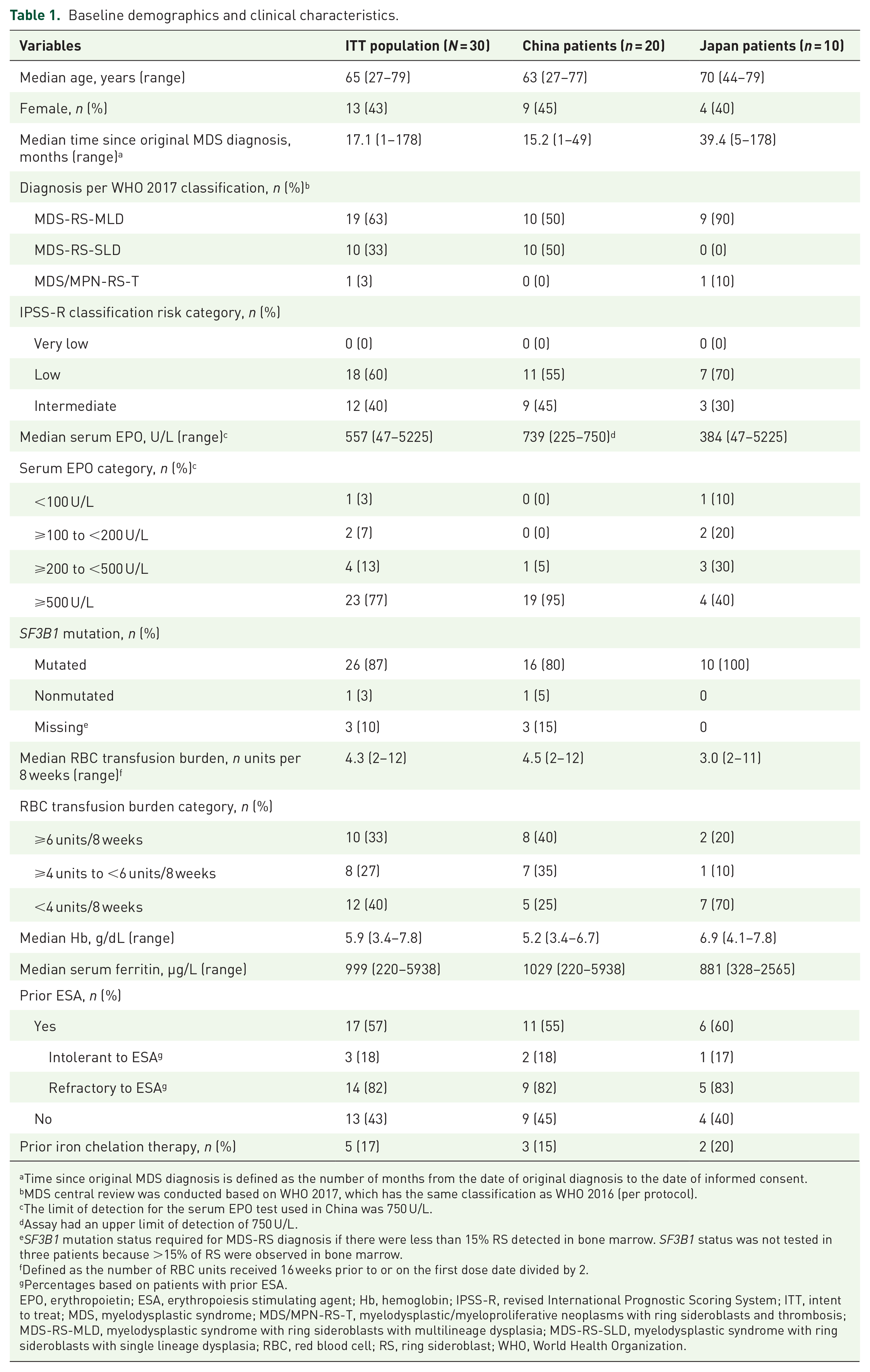
Between November 2020 and March 2023, 37 patients were screened, 30 of whom were enrolled in the study (ITT population). In total, 20 Chinese patients and 10 Japanese patients were enrolled. Among the ITT population, most patients were male (57%) and had a median age of 65 years (range, 27–79 years; Table 1).
Baseline demographics and clinical characteristics.
Time since original MDS diagnosis is defined as the number of months from the date of original diagnosis to the date of informed consent.
MDS central review was conducted based on WHO 2017, which has the same classification as WHO 2016 (per protocol).
The limit of detection for the serum EPO test used in China was 750 U/L.
Assay had an upper limit of detection of 750 U/L.
SF3B1 mutation status required for MDS-RS diagnosis if there were less than 15% RS detected in bone marrow. SF3B1 status was not tested in three patients because >15% of RS were observed in bone marrow.
Defined as the number of RBC units received 16 weeks prior to or on the first dose date divided by 2.
Percentages based on patients with prior ESA.
EPO, erythropoietin; ESA, erythropoiesis stimulating agent; Hb, hemoglobin; IPSS-R, revised International Prognostic Scoring System; ITT, intent to treat; MDS, myelodysplastic syndrome; MDS/MPN-RS-T, myelodysplastic/myeloproliferative neoplasms with ring sideroblasts and thrombosis; MDS-RS-MLD, myelodysplastic syndrome with ring sideroblasts with multilineage dysplasia; MDS-RS-SLD, myelodysplastic syndrome with ring sideroblasts with single lineage dysplasia; RBC, red blood cell; RS, ring sideroblast; WHO, World Health Organization.
All patients had MDS diagnosis per WHO 2017 classification: 63% as MDS-RS with multilineage dysplasia (MDS-RS-MLD), 33.3% as MDS-RS with single-lineage dysplasia (MDS-RS-SLD), and 3.3% as MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T), assessed by central laboratory. Per IPSS-R criteria at enrollment, 60% of patients had low risk, and 40% of patients had intermediate-risk MDS (Table 1). Median baseline Hb was 5.85 g/dL, and all patients had Hb under 8 g/dL. Median baseline serum ferritin was 999.1 μg/L, which was consistent with historical serum ferritin levels (defined as average serum ferritin value within 16 weeks prior to first dose; median: 925.9 μg/L). In total, 23.3% of patients (7/30) had a serum EPO ⩽500 U/L at baseline, while 76.7% of patients (23/30) had a serum EPO > 500 U/L (Table 1).
The median RBC transfusion burden per 8 weeks during the 16 weeks prior to the first dose was 4.25 units (range, 2–12). 33.3% of patients required ⩾6 units, 26.7% required ⩾4 to <6 units, and 40% required <4 units (Table 1). A total of 13 (43.3%) patients were ESA naïve.
The clinical data cutoff date was September 29, 2023. Median (range) follow-up (time from the first dose date to the clinical data cutoff date) was 12.2 (1.5–32.1) months. Nineteen patients (63.3%), 12 from China and 7 from Japan, discontinued from the treatment, and the most common reason was lack of efficacy (7 patients, 23.3%). Six patients (20.0%) discontinued from the study as of the clinical data cutoff date, and the most common reason was withdrawal by patient (three patients, 10%). The majority of discontinuations occurred after the 24-week assessment.
Exposure
All enrolled patients received at least one dose of luspatercept. The median (range) duration of treatment was 32.9 (3–127) weeks, and the median (range) number of doses was 11.5 (1–42) per patient. Twenty-four patients (80%) completed 24 weeks of treatment, and nine patients (30%) completed 48 weeks of treatment. At the clinical data cutoff date, 11 (37%) patients continued to receive luspatercept. In total, 22 (73%) patients had at least one dose titration of luspatercept; 22 (73%) patients escalated from 1.0 to 1.33 mg/kg; and 18 (60%) patients escalated from 1.33 to 1.75 mg/kg. Overall, 8 (27%) patients received a maximum dose of 1.0 mg/kg luspatercept, 4 (13%) received a maximum dose of 1.33 mg/kg, and 18 (60%) received a maximum dose level of 1.75 mg/kg.
Efficacy
In the ITT population, a statistically significant and clinically meaningful improvement was observed in the primary endpoint, with 18 patients (60.0%) achieving RBC-TI ⩾8 weeks from week 1 through week 24 (p < 0.0001; Table 2). The swimmer plot of transfusion events and RBC-TI ⩾8 weeks response during entire treatment phase for each patient is provided in Supplemental Figure 1. By Kaplan–Meier estimates, the median duration of RBC-TI ⩾8 weeks (week 1 through EOT) was 24.43 weeks (95% CI: 15.1–81.9; Supplemental Figure 2). Among the patients who achieved RBC-TI ⩾8 weeks from week 1 through week 24, there was a rapid onset of effect with luspatercept, with response achieved in a median (range) of 1.0 (1–93) day after the first dose (Supplemental Table 3).
Rates of RBC-TI.

Exact test was performed to compare the response rate with 13%, which was obtained from MEDALIST results.
CI, confidence interval; ITT, intent to treat; RBC-TI, red blood cell transfusion independence.
In total, 13 patients (43.3%) achieved RBC-TI ⩾12 weeks from week 1 through week 24 (Table 2). The median baseline RBC transfusion burden over the 16 weeks immediately preceding the first luspatercept dose was 8.50 units in ITT population (Supplemental Table 4). During weeks 9–24 of treatment, there was a 47% decrease in transfusion burden to 3.0 units. During weeks 1–24, 21 (77.8%) and 14 (53.8%) patients experienced a maximum transfusion burden reduction of ⩾50% in any 8- and 16-week period, respectively (Figure 1).

Waterfall plots of maximum RBC transfusion reduction, (a) ⩾8 weeks (week 1–24)a and (b) ⩾16 weeks (weeks 1–24)b (ITT population).
In the exploratory endpoint of reduction in RBC units transfused over 24 weeks compared with baseline (China patients only), the median baseline RBC transfusion burden over the 24 weeks immediately preceding or on the first luspatercept dose was 13.0 units. During weeks 1–24 of treatment, there was a ~58% decrease in transfusion burden to 5.5 units. In the exploratory endpoint of mean Hb change over 24 weeks (China patients only), the mean Hb in the 24 weeks immediately preceding or on the first luspatercept dose date was 5.6 g/dL (SD, 0.9 g/dL), which increased to 6.6 g/dL (SD, 1.6 g/dL) over weeks 1–24.
In total, 19 (63.3%) patients achieved mHI-E per IWG criteria (2006) between weeks 1 and 24 (Table 3). Sixteen patients (53.3%) had a mean Hb increase of ⩾1.0 g/dL from week 1 to week 24 (Table 3). The mean change from baseline in mean serum ferritin during weeks 9–24 was −48.5 µg/L (SD, 980.3 µg/L).
Erythroid response and increase in hemoglobin levels.

CI, confidence interval; ITT, intent to treat; IWG, International Working Group; mHI-E, modified hematologic improvement–erythroid.
At data cutoff, no patient had acute myeloid leukemia progression. Two patients (6.7%) died in the posttreatment period, one due to malignant disease under study (or complications due to malignant disease under study), and one due to a cardiac event. Neither death was considered to be related to study treatment. Median follow-up time for OS was 12.19 months, and median OS was not reached (Supplemental Figure 1).
Exploratory subgroup analysis
In an exploratory analysis of efficacy endpoints by baseline characteristic subgroup, consistent response rates were observed in ITT subgroups with ⩾10 patients. Rates of RBC-TI ⩾8 weeks (weeks 1–24) were generally consistent between those who were ESA naïve (n = 13) and ESA refractory/intolerant (n = 17), with 69% (95% CI: 39–91) and 53% (95% CI: 28–77) of patients responding, respectively.
Pharmacokinetics
In patients with no dose modification, one dose escalation, and two dose escalations, mean trough luspatercept serum concentration was generally stable after reaching steady state (Supplemental Figure 3). The steady-state concentration seemed to be slightly higher in patients with two dose escalations, though no definite conclusions can be drawn due to low patient numbers.
Safety
A total of 28 patients (93.3%) reported at least one treatment-emergent adverse event (TEAE), the most common of which was COVID-19 (33.3%; Table 4). Ten patients (33.3%) reported at least one grade 3/4 TEAE, the most common of which were COVID-19 pneumonia, iron overload, and neutropenia (6.7% each). Eight patients (26.7%) reported at least one serious TEAE; two (6.7%) had a serious TEAE considered by the investigator to be related to luspatercept. Two (6.7%), six (20.0%), and one (3.3%) patient reported a TEAE leading to luspatercept discontinuation, dose interruption, and dose reduction, respectively. No patients died while on treatment.
Safety summary.
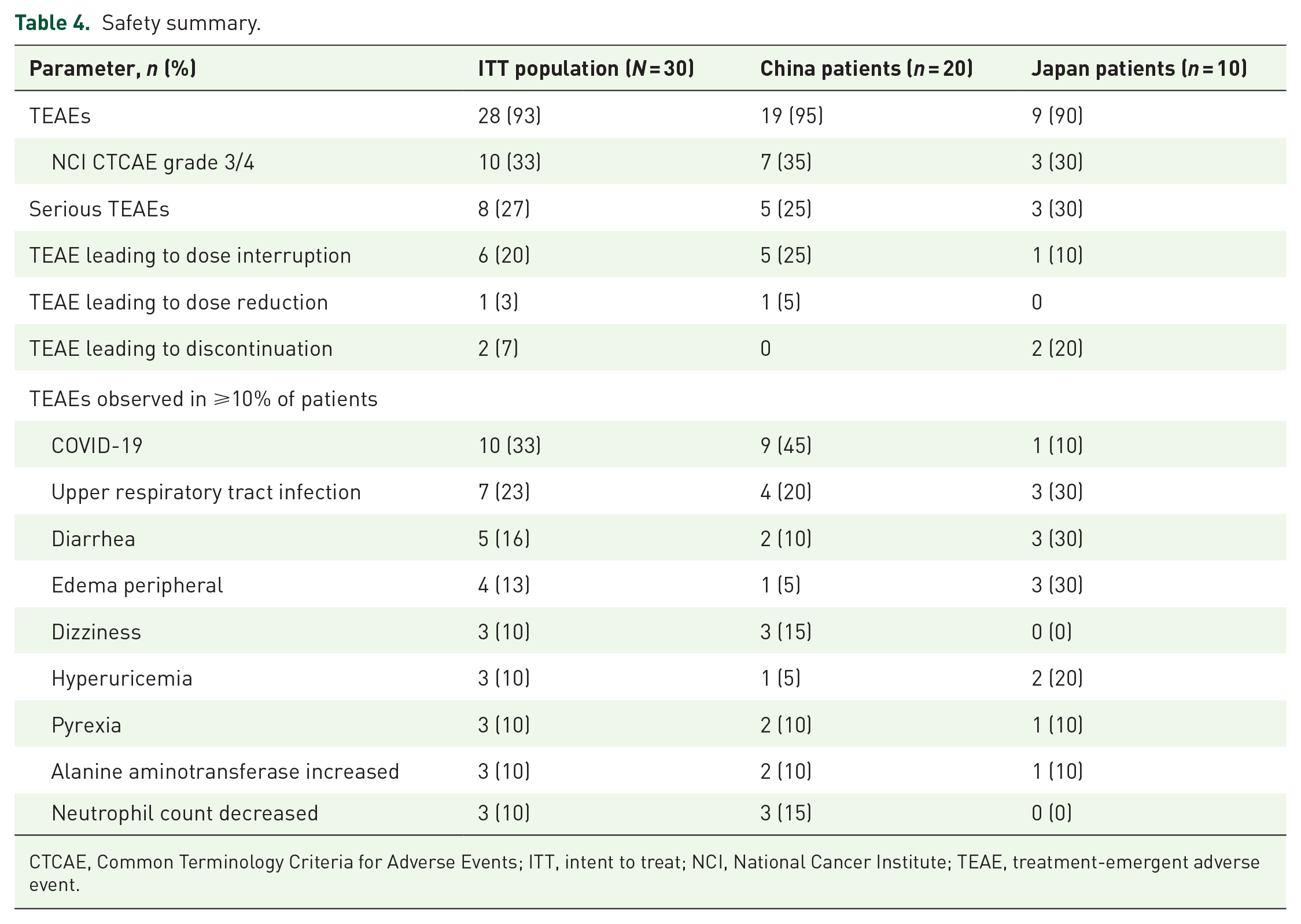
CTCAE, Common Terminology Criteria for Adverse Events; ITT, intent to treat; NCI, National Cancer Institute; TEAE, treatment-emergent adverse event.
Discussion
This single-arm, phase II bridging study assessed the efficacy, safety, and PK of luspatercept in an Asian population (China and Japan) with non-del(5q) LR MDS-RS who requires RBC transfusions for anemia and who were refractory, intolerant, or ineligible to receive an ESA. The study design and population reflected that of the phase III MEDALIST study (conducted in an ex-Asian population), in order to assess the safety and efficacy of luspatercept in a representative Asian population.
The MDS population enrolled into this study is representative of the transfusion-dependent LR-MDS-RS population in China and Japan, where patients have a highly symptomatic disease (with low Hb level and high serum EPO level, requiring regular transfusions) and, once ESAs are no longer effective, have limited therapeutic options beyond supportive care. Baseline characteristics were generally balanced between this bridging study and the pivotal MEDALIST study, but with some important differences. These were largely representative of differences in patients with MDS between Asia and North America/Europe and likely due to variations in treatment approaches in the respective countries. In the current study, a higher proportion of patients received luspatercept as first-line treatment, since they were ESA naïve (patients not previously treated with ESAs) due to endogenous serum EPO level >200 U/L and therefore were ESA ineligible per protocol compared with the MEDALIST study. Additionally, patients had a lower median age, a shorter time since original MDS diagnosis, a lower proportion of MDS-RS-MLD, a higher proportion with baseline EPO >500 U/L, and lower median Hb level versus the MEDALIST study patients. Similar differences in clinical features between Asian and Western MDS patient populations have been identified in other studies. 25
This phase II bridging study met its primary endpoint; luspatercept was effective in achieving statistically significant RBC-TI for ⩾8 weeks (weeks 1–24) in 60% (n = 18) of this population. Notwithstanding differences in clinical features between the trial populations, 21 RBC-TI ⩾8 weeks (weeks 1–24) was achieved in 38% of luspatercept-treated patients versus 13% in placebo group (p < 0.001) in the MEDALIST study, which is in line with the current study. Results for the secondary endpoints were also similar between the two studies. The response to luspatercept was rapid and durable, as the start of the RBC-TI ⩾8 weeks response occurred in a median of 1.0 day from week 1 though week 24. This response profile closely parallels the findings from the MEDALIST study, where 43.3% of patients achieved RBC-TI ⩾12 weeks from week 1 through week 24 compared with 28.1% in this study.
Among all patients in the ITT population, there was approximately a 65% reduction in the median number of RBC units transfused over a fixed 16-week interval, decreasing from 8.5 units at baseline to 3 units during the primary phase (assessed between weeks 9 and 24). These results were in line with the MEDALIST study, where there was a 40% decline from 10.0 to 6.0 units. Further, 53.3% (16/30) of patients achieved mean Hb increase of ⩾1.0 g/dL during weeks 1 through 24, which is also in line with the 35.3% observed in the MEDALIST study.
Of those treated with luspatercept, 63.3% achieved mHI-E over a consecutive 8-week period. In MEDALIST, 52.9% of luspatercept-treated patients achieved mHI-E. In patients with a baseline transfusion burden ⩾4 units/8 weeks, 61.1% experienced a reduction in RBC transfusion of >4 units over a consecutive 8-week period from baseline during weeks 1 through 24, compared with 48.6% in MEDALIST. In those with a baseline transfusion burden <4 units/8 weeks, 66.7% had a mean Hb increase of ⩾1.5 g/dL from baseline, compared with 63.0% in MEDALIST.
Safety and tolerability parameters were comparable between the two studies. Luspatercept had an acceptable safety profile with most TEAEs classified as grade 1/2. Common TEAEs included edema and fatigue. PK parameters were also comparable between the two studies.
This study was limited by the small number of patients, the lack of blinding, and the lack of a comparator group. Additionally, direct comparisons between this phase II bridging study and the MEDALIST trial should be interpreted with caution, due to differences in sample size and patient baseline clinical features, which may have been confounding.
During the 24 weeks of treatment, luspatercept demonstrated durable and clinically meaningful benefit. These results are promising in an Asian clinical context, given that a large part of this cohort had high EPO levels, which is representative of transfusion-dependent lower-risk MDS (TD-LR-MDS) patient populations in China and in Japan. These high EPO levels are likely due to a compensatory response in the context of severe anemia in these populations, particularly in China, where there is limited availability of blood for transfusion.28,29
In addition to the results from the MEDALIST and this phase II bridging study on luspatercept as a treatment option for TD-LR-MDS in the second-line setting, recent publications from the COMMANDS clinical trial have further demonstrated the clinically meaningful benefits of luspatercept across multiple subgroups including RS-positive and RS-negative patients, as well as those with mutated and nonmutated SF3B1 TD-LR-MDS. 23 These findings highlight the potential of luspatercept as a first-line treatment worldwide, including in Asian populations, and offer a promising approach to early intervention in MDS that could lead to improved patient outcomes.
Conclusion
Luspatercept had an acceptable and manageable safety profile that was consistent with the known safety profile in patients with MDS. These results suggest that luspatercept can provide a well-tolerated and efficacious alternative to transfusions for Asian patients with transfusion-dependent LR-MDS-RS and align with the results observed in the global MDS population reflected in the pivotal phase III MEDALIST trial.
Supplemental Material
sj-docx-1-tah-10.1177_20406207251321715 – Supplemental material for Safety and efficacy of luspatercept in treating anemia associated with myelodysplastic syndrome with ring sideroblasts in Asian patients who require red blood cell transfusions: a phase II bridging study
Supplemental material, sj-docx-1-tah-10.1177_20406207251321715 for Safety and efficacy of luspatercept in treating anemia associated with myelodysplastic syndrome with ring sideroblasts in Asian patients who require red blood cell transfusions: a phase II bridging study by Chunkang Chang, Takahiro Suzuki, Yang Liang, Hongyan Tong, Kensuke Usuki, Qifa Liu, Yu Wu, Tomoaki Fujisaki, Bing Han, Ruibin Huang, Yasuyoshi Morita, Miao Miao, Yasuhiro Nakashima, Yu (Olivia) Tian, Jie Pu, Dimple Aggarwal, Veronika Pozharskaya, Wenhui Shi, Zhijian Xiao and Kinuko Mitani in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
We thank all patients and families who participated in the trial. Ana Carolina Giuseppi contributed to trial design. Editorial and writing assistance were provided by Christopher Spencer, PhD, of Parexel, funded by Bristol Myers Squibb.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.