
Abstract
Children with Down syndrome (DS) are at increased risk of developing haematological malignancies, in particular acute megakaryoblastic leukaemia and acute lymphoblastic leukaemia. The microenvironment established by abnormal haematopoiesis driven by trisomy 21 is compounded by additional genetic and epigenetic changes that can drive leukaemogenesis in patients with DS. GATA-binding protein 1 (GATA1) somatic mutations are implicated in the development of transient abnormal myelopoiesis and the progression to myeloid leukaemia of DS (ML-DS) and provide a model of the multi-step process of leukaemogenesis in DS. This review summarises key genetic drivers for the development of leukaemia in patients with DS, the biology and treatment of ML-DS and DS-associated acute lymphoblastic leukaemia, late effects of treatments for DS-leukaemias and the focus for future targeted therapy.
Keywords
Introduction
Down syndrome (DS) is characterised by constitutional acquisition of a partial or complete third copy of chromosome 21. It is the most common single chromosomal disorder in live births, with an incidence of 1 in 300 to 1 in 1100 babies. 1 DS is associated with several haematological abnormalities. Patients with DS are at particular risk of developing acute megakaryoblastic leukaemia (AMKL) (500-fold risk compared to the non-DS population) and acute lymphoblastic leukaemia (ALL) (20-fold risk). 1 Unique to children with DS or mosaic trisomy 21 (T21) is the development of transient abnormal myelopoiesis (TAM), a preleukaemic state which develops in the prenatal period and increases the risk of development of myeloid leukaemia of DS (ML-DS).
This review will focus on recent developments in the understanding of ML-DS and DS-associated ALL (DS-ALL) and clinical updates in the management of these conditions, including therapies to limit treatment-associated toxicity and late effects of treatment. It will provide insights into clinically relevant future focuses for research in leukaemogenesis and potential targets for therapeutic advances in patients with DS-associated leukaemias.
Role of T21 in leukaemogenesis
The development of leukaemia in DS is a multi-step process initiated by the intrinsic chromosomal abnormality of T21 (Figure 1). T21 induces an imbalance in haematopoietic stem and progenitor cell (HSPC) differentiation during foetal liver haematopoiesis. This results in multipotent progenitor cells being preferentially directed towards erythroid or erythro-megakaryoblastic differentiation and subsequent expansion of erythroid and megakaryocytic progenitors, at the expense of terminal B-cell differentiation and natural killer (NK) lineage output.2–6

Overview of leukaemogenesis in Down syndrome.
Individuals with T21 demonstrate presence of clonal haematopoiesis at higher frequency and younger age compared to disomic individuals. 7 Haematopoetic stem cells, differentiated lymphocytes and mononuclear cells from individuals with T21 display defective DNA repair mechanisms and increased sensitivity to oxidative damage, contributing to a state of genomic instability.8,9 Increased expression of DYRK1A, located on chromosome 21, in haematopoietic cells caused significant derangement of DNA damage repair pathways in T21-derived induced pluripotent stem cell (iPSC) models, giving a possible mechanism for genome instability in T21.10,11 In this way, T21 provides a microenvironment conducive to expansion of clonal populations and the acquisition of oncogenic mutations. 7 Single-cell studies have demonstrated increased mutagenesis in T21 foetal haematopoietic stem cells, leading to an increased risk of acquiring additional leukaemogenic mutations. 12
Abnormal methylation induced by T21 has been implicated in dysregulated haematopoiesis. Muskens et al. 13 analysed neonatal bloodspots from 196 newborns with DS and 439 newborns without DS and identified differential methylation at several promoter/enhancer regions, including the RUNX1 and FLI1 regions that are known to regulate haematopoietic development. Methylation changes were found not just in chromosome 21 but across the genome, indicating the impact of T21 on genome-wide gene regulation. 13
Polysomy of chromosome 21 is one of the most frequent chromosomal abnormalities seen in haematological malignancies and is suggestive that leukaemogenesis may be driven by megakaryocytes and B-cell precursors that are more sensitive to increased gene dosage. 14 Somatic T21 is seen in approximately one-third of children with pre-B-ALL and AMKL. 14 Intra-chromosomal amplification of chromosome 21 (iAMP21), a cytogenetic indicator of high-risk (HR) disease, is seen in 2% of children with B-ALL. 15 iAMP21 is characterised by areas of amplification, particularly the region containing the DS critical region (DSCR) and RUNX1, 14 indicating that this chromosomal region may play a role in leukaemogenesis.
Interestingly, patients with DS have a lower risk of solid tumours overall compared to the general population for reasons that are unclear. 16 Some groups have suggested that DS represents a ‘leukaemogenic’ state due to the possible genetic instability that constitutional T21 carries rather than a cancer predisposition syndrome.16,17 There is a suggestion, however, that within the solid tumours that are recorded in patients with DS, different patterns of location and subtypes of tumour have been observed compared to the general population, with an increase in testicular cancer for example in adolescents and young men with DS. 18 A specific example in children with DS is that germ cell tumours make up a disproportionate number of central nervous system tumours, with a higher percentage located in the basal ganglia, compared to the non-DS population. 19
The diversity of phenotypes in patients with DS-associated leukaemias is postulated to be due to a number of mechanisms. These hypotheses include: (i) increased gene expression of human chromosome 21 (Hsa21)-based genes that are dosage sensitive, thereby contributing to the phenotype; (ii) the amplified developmental instability hypothesis, which suggests that genetic imbalance is caused by trisomic genes impacting the expression and regulation of many genes; and (iii) the critical region hypothesis, whereby specific genes on Hsa21 form the DSCR, which is responsible for the typical phenotypic features of patients with DS. 20 Several genes which have been found to be associated with leukaemia are located in the DSCR, including ERG, ETS2, DYKR1A and RUNX1.21–24 It has been proposed that overexpression of these genes in the DSCR induces aberrant megakaryopoiesis.25,26
Germline cancer predisposition genes may also interact with T21. Whilst the incidence of pathological germline predisposition variants in children with DS-associated ALL (DS-ALL) versus non-DS-ALL is similar, T21 may modify and increase the penetrance of several germline variants in ALL predisposition genes, including IKZF1, GATA3 and CDKN2A.27,28
An understanding of the complex interplay between the T21 microenvironment and additional acquisition of leukaemogenic mutations provides a basis for understanding the biology of TAM, ML-DS and DS-ALL, as well as future targets for therapies.
Myeloid leukaemia of DS
The multi-step leukaemogenesis model of disease is demonstrated by TAM, a preleukaemia that develops in utero. In 20–30% of cases, this later develops into ML-DS.29,30 TAM is also referred to as transient myeloproliferative disorder (TMD) or transient leukaemia (TL). The embryonal origin of childhood cancer has been well researched for TAM/ML-DS, as well as other malignancies, including ALL and medulloblastoma. 31
ML-DS presents with a unique, clonally-related, self-limiting neonatal preleukaemic syndrome that is characterised by the presence of circulating megakaryoblasts and somatic GATA1 mutation.32,33 The reported incidence of TAM ranges from 5% to 30% of liveborn children with T21.33,34 TAM presents antenatally or in the early neonatal period with a wide spectrum of clinical severity, from clinically silent disease to rapidly fatal multiorgan failure due to leukaemic infiltration, which has a mortality up to 20%.30,32–35 TAM spontaneously resolves within 3–4 months of life in 80–90% of cases.32,33,35 Altered signalling and growth factors in the foetal liver haematopoietic niche in infants with DS play a critical role in survival and proliferation of TAM cells.36,37 Transition to bone marrow haematopoiesis and the loss of the supportive environmental niche may account for the spontaneous resolution of TAM in many cases.36,37 In the 20–30% of children with TAM who go on to develop ML-DS, progression to ML-DS occurs within the first 4 years of life when persistent GATA1 mutant cells acquire additional driver mutations triggering leukaemic transformation.30,32,33,35
Molecular drivers in ML-DS
Acquisition of a GATA1 mutation and the synergistic interaction between T21 and GATA1 mutations located on the X chromosome represent key steps in ML-DS pathogenesis. The GATA1 transcription factor plays a key role in the regulation of haematopoietic stem cell development and differentiation, particularly of erythroid and megakaryoblastic lineages.32,38 Acquisition of one or more N-terminal mutations in exon 2 or exon 3 of the GATA1 gene results in loss of full-length GATA1 (GATAfl) expression and exclusive translation of the short isoform of GATA1 (GATA1s).39,40 The exact mechanism by which T21 drives acquisition of GATA1 mutations remains unknown. The presence of both constitutional T21 and acquired GATA1s drives uncontrolled expansion of megakaryocytes and perturbed terminal erythroid differentiation, further driving expansion of the megakaryocyte-erythroid compartment and accumulation of clonal populations.32,37,41 GATA1s amplifies genome instability seen in constitutional T21 by further upregulating DYRK1A expression, impeding double-stranded DNA repair pathways and augmenting accumulation of chromosomal aberrations. 10 Isolated germline GATA1 mutations are not associated with leukaemia in the absence of T21; instead, presenting with failure of terminal differentiation of erythropoiesis and anaemia. 42
Molecular drivers implicated in leukaemogenesis in ML-DS include genes within the DSCR (RUNX1, DYRK1A, ERG and ETS2), cohesin complex genes (STAG1, STAG2), microRNAs, methylation changes and the wider epigenetic impact of T21 on haematopoiesis.
The DSCR contains genes on chromosome 21 that are responsible for the differing phenotypes in DS. A segment (estimated to be ~4Mb – 8.35Mb) containing genes responsible for haematopoietic differentiation, including RUNX1, ERG and ETS2, has been identified within the DSCR and has been found to play a role in leukaemogenesis in DS.26,43 Comparison of iPSCs with trisomy of the DSCR to partial trisomy 21 IPSCs (with deletion of the ~4Mb critical segment) found that this region is essential in driving haematopoiesis in T21, and in contributing to perturbed megakaryocyte development. 26 T21-induced RUNX1 isoform disequilibrium and RUNX1A isoform overexpression synergises with GATA1s to drive proliferation and accumulation of immature megakaryocytic progenitors. 44 ERG, ETS2 and FLI1 overexpression has been associated with immortalisation of foetal liver progenitors cells in GATA1 knockdown and GATA1s knock-in murine cell lines. 24 In these mouse models there was also activation of the Janus kinase (JAK)/signal transducer and activation of transcription proteins (STAT) (JAK-STAT) pathway signalling. 24 The JAK-STAT pathway has been identified as a therapeutic target, with use of JAK2 inhibitors a potential avenue to advance management of both ML-DS and DS-ALL.45,46
Mutations in cohesin complex genes have been implicated in the multi-step process of leukaemogenesis, after acquisition of GATA1 mutations. 32 Cohesin is a multimeric protein complex that wraps around DNA, forming a ring-like structure that is critical for three-dimensional architecture and structural genome organisation, allowing for dynamic changes in genome expression and transcriptional activation. 47 Cohesin-mediated changes in chromatin accessibility alter differential binding of self-renewal and differentiation transcriptional factors.47,48 Cohesin is also essential for the congregation and segregation of sister chromatids and for maintaining DNA integrity and repair. 47 The key cohesin complex genes involved in transcription, including RAD21, STAG2, SMC3 and SMC1A, account for 53% of mutations in patients with ML-DS. 49 The frequency of cohesin mutations is higher in ML-DS compared to AML in children without DS. 47 In ex vivo and mouse models, cohesin is necessary to maintain haematopoietic differentiation.47,50,51 Deficiency or overexpression of cohesin in human haematopoietic stem cells can lead to increased self-renewal capacity and alterations in differentiation and lineage commitment of haematopoietic and progenitor stem cells, which can result in inappropriate activation of haematopoietic enhancers.52,53 CCCTC-binding factor (CTCF) co-localises and interacts with cohesin to regulate the three-dimensional architecture of chromatin and regulation of gene expression by arresting the DNA loop extrusion activity of cohesin.47,48,54 In mouse models, CTCF appears to play an important role in erythroid growth and differentiation. 54 Loss-of-function mutations in CTCF are present in 2% of patients with TAM and 20% of patients with ML-DS, 49 and may play a role in progression of TAM to ML-DS. 49
Overexpression of microRNAs located on chromosome 21, mainly miR-99a, miR-125b-2, miR-155, have been found to increase myeloid and decrease lymphoid differentiation in the bone marrow of mouse models. 55 Klusmann et al. 56 demonstrated that miR-125b-2 plays an important role in megakaryopoiesis, and overexpression of this microRNA can perturb myeloid differentiation. Furthermore, there is a synergistic effect of miR-125b-2 and GATA1 mutations on proliferation of the megakaryocytic/erythroid progenitor compartment and differentiation arrest through miR-125b-mediated repression of the megakaryocytic transcription factor ARID3A, implicating microRNAs in the development of ML-DS. 57
Models of ML-DS leukaemogenesis
One of the most extensively studied mouse models for DS is Ts65Dn, which has 104 orthologs of Hsa21. 58 Ts65Dn mice demonstrate phenotypic features of DS with distinctive craniofacial features, cognitive impairment and cardiac defects. CD34+ cells isolated from Ts65Dn mice showed decreased proliferation compared to CD34+ cells isolated from diploid mice, with a relative increase in presence of the TP53 protein. 59 Ts65Dn mice develop a myeloproliferative disease by 15 months of age, associated with thrombocytosis, extramedullary haematopoiesis, and distorted stem and myeloid progenitor cell compartments. 60 However, leukaemia has not been reported in these mice, indicating that there are other factors involved in leukaemogenesis.
A double transgenic mouse model expressing Gata1s and ETS transcription factor ERG (located in the DSCR) has been developed, 61 which demonstrates that GATA1s expression works synergistically with the oncogene ERG to expand the megakaryocyte/erythroid progenitor compartment in foetal livers. This model developed clinical features consistent with TAM, with thrombocytosis and hepatic fibrosis, and progression to megakaryoblastic-erythroid leukaemia. 61 Most male mice with ERG transgene/GATA1s double expression did not survive beyond day 12 of embryogenesis due to anaemia. 61 Ng et al. 21 demonstrated in the Ts(17 16 )65Dn mouse model that functional disomic correction of ERG corrects the abnormal myeloproliferation seen in the trisomy mice. While these in vivo models have enhanced our understanding of critical gene dosage required for myeloproliferation and leukaemogenesis in DS, it remains a challenge to develop a model to study the complex interactions between T21, genetic and epigenetic drivers that are involved in the development of TAM and ML-DS. 62
iPSCs provide another option to model leukaemogenesis. Barwe et al. 63 studied the impact of STAG2 knockout mutations on iPSCs with GATA1 mutations and found that the double mutant HSPCs cooperatively increased the megakaryocyte population and increased expression of ML-DS markers, impacting on megakaryocyte differentiation. Using iPSCs, Banno et al. 26 used differentiation experiments to demonstrate abnormal haematopoiesis in T21, and that acquired GATA1s mutation leads to aberrant megakaryoblast development driven by RUNX1/ETS2/ERG-mediated and GATA1s dose-dependent hyperproliferation.
Increasingly, model systems are using novel methods such as CRISPR-Cas9 gene editing. Analysis of loss-of-function mutations with CRISPR-Cas9 screening of clinical TAM and ML-DS samples has identified 18 genes that cooperate with GATA1 mutations in the abnormal haematopoietic landscape of T21. 64 Labuhn et al. 64 identified that, in a population of 252 clinical samples, the combination of T21 and GATA1 mutations was sufficient to result in TAM, while in the majority of ML-DS samples, additional somatic variants were acquired.
Clinical features of TAM and ML-DS
Almost all neonates with DS have multiple quantitative and morphologic haematological abnormalities at birth, including presence of blasts, and therefore haematological criteria alone are not reliably diagnostic of TAM nor predictive of development of ML-DS33,34 (Figure 2). Megakaryoblastic TAM cells spread from their origin in the foetal liver, to infiltrate throughout the liver and into the peripheral blood and skin, causing characteristic features of TAM including hepatomegaly, coagulopathy, papular or vesicopustular skin rash and pericardial and pleural effusions.30,33,34 The presence of blasts in bone marrow is variable and less prevalent than in peripheral blood. 35 Bone marrow involvement does not correlate with disease severity.29,30,35 This clinical observation further supports the importance of the foetal liver haematopoietic compartment in pathogenesis of TAM. Splenomegaly occurs in 30% of neonates with TAM, predominantly due to portal venous obstruction, as splenic infiltration is rarely seen. 33
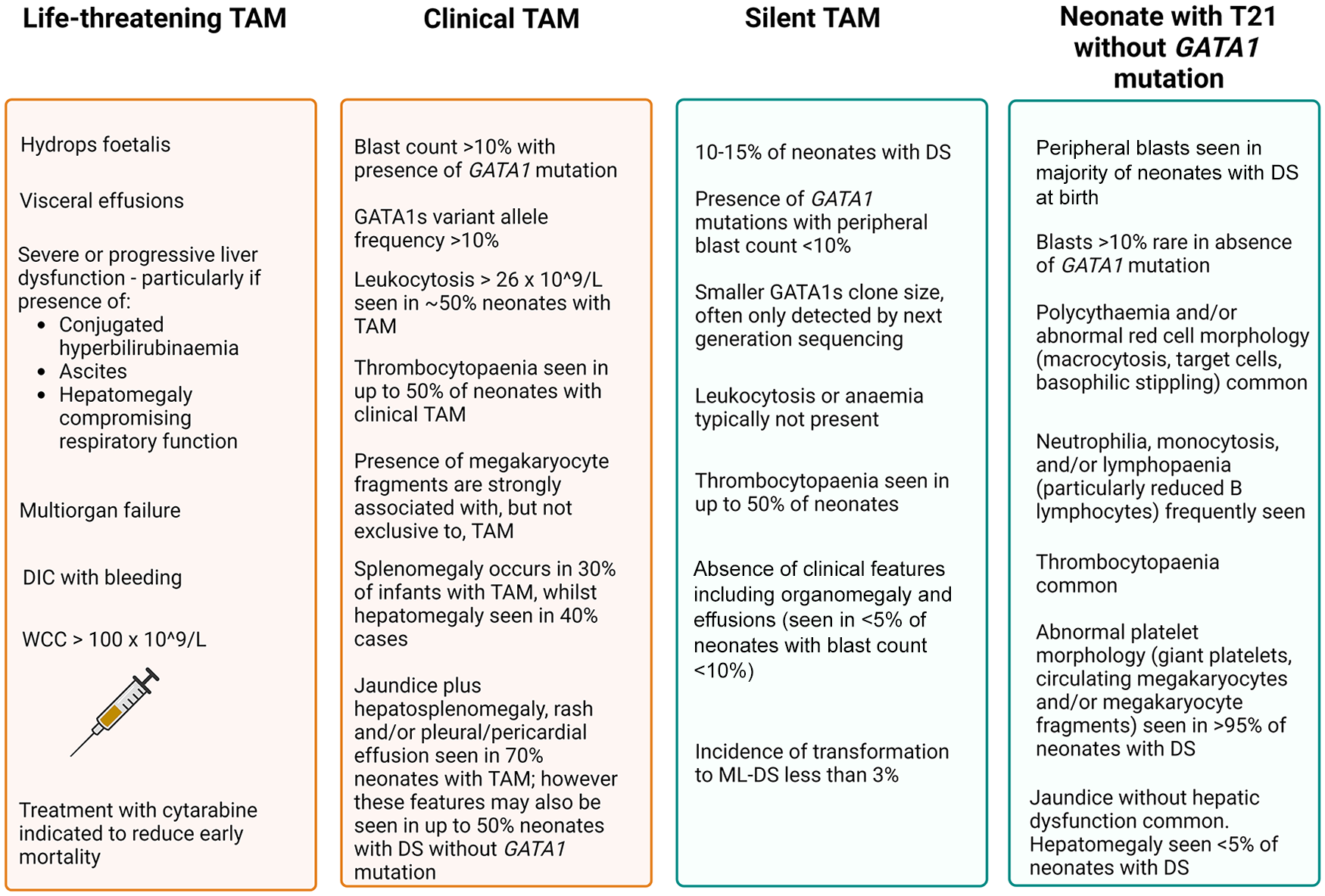
Summary of clinical and haematological features of TAM.
Prospective studies of neonates with DS have demonstrated that the presence of mutant GATA1s clones is an independent predictive factor for the development of ML-DS. ML-DS did not occur in any infant without prior presence of a GATA1s mutation, irrespective of blast count at birth.65,66 There is uncertainty regarding the minimum percentage of blasts required to make a diagnosis of TAM, as infants with DS are more likely to have immature circulating cells in peripheral blood at birth even in the absence of GATA1s mutation. 33 Goemans et al. 66 screened a population of Dutch infants for TAM as a predictor for the development of ML-DS. Their recommendation was to define TAM as >5% blasts identified by immunophenotyping or morphology and/or the presence of a GATA1 mutation in an infant with DS to sufficiently identify infants at risk of developing ML-DS, 66 while Roberts et al. 34 have proposed a >10% threshold. GATA1 mutant clones have been detected in 10–15% of neonates with DS in the absence of circulating blasts or clinical symptoms, 34 referred to as ‘silent TAM’. Critically, transformation to ML-DS may occur in children with silent TAM, although the incidence is less than 3%, lower than the 30% incidence of development of ML-DS in infants with clinical TAM.30,34
Interestingly, there are rare reported cases of patients who have developed DS-ALL following previous diagnosis of TAM or ML-DS. 67 Cytogenetic data, including presence or absence of GATA1 in DS-ALL clones, in these cases is limited, and it is unclear whether these cases are evidence of a common stem cell that can give rise to either myeloid or lymphoid lineage leukaemia, or reflective of increased risk of both types of leukaemia in children with DS.
To date, there have been no cytogenetic abnormalities identified that are reliably predictive of development of ML-DS.29,30 There is conflicting evidence regarding whether GATA1-related factors predict development of ML-DS. One study found that the type of GATA1 mutation type was not prognostic, 68 while another study determined that GATA1s protein levels were prognostic of subsequent ML-DS progression. 69 Detectable minimal residual disease (MRD) by flow cytometry at 12 weeks of age in infants with TAM is a significant predictive indicator for development of ML-DS, as is persistent MRD by PCR at 12 weeks after TAM diagnosis.70,71 The value of monitoring for persistence of GATA1 mutant clones beyond 3 months after TAM diagnosis, by PCR or flow cytometry, has not yet been reported. Up to 25% of cases of TAM display multiple mutant GATA1 clones34,49,68,72 and ML-DS may develop from a minor GATA1-mutant clone, 49 adding to the complexity of MRD surveillance.
ML-DS typically presents by 4 years of age, with the median age of onset being 12–18 months.32,38 AML may occur in children with DS over the age of 4 years; however, it appears to represent a separate disease process, with lack of GATA1 mutations and cytogenetic findings more in keeping with sporadic AML seen in children without DS. 73 ML-DS is often preceded by pancytopenia with dysplastic marrow morphology and presents with a relatively low circulating blast cell count.74,75 Paired TAM and ML-DS samples show presence of the same GATA1 mutation, demonstrating that these are clonally linked.68,72 ML-DS is characterised by a distinct immunophenotype and cytogenetic changes that differ from non-DS AML, with absence of common chromosomal translocations and inversions commonly seen in non-DS AML, such as inv(16) and t(8;21).75,76 Trisomy 8, tetrasomy 21, gain of 1q and loss of 7p have been identified as the most frequent cytogenetic abnormalities in ML-DS.76,77
Treatment of TAM and ML-DS
Spontaneous remission without sequelae occurs in most cases of TAM, with overall survival (OS) of 85% with supportive care alone.29,30,35 Early mortality before 6 months of age due to TAM occurs in 15–20% infants.29,30,33,35,78 The predominant causes of early death are progressive liver dysfunction leading to fulminant hepatic fibrosis and disseminated intravascular coagulopathy (DIC) and multiorgan failure, or progressive pleural/pericardial effusions resulting in cardiorespiratory failure.29,30,78 Independent risk factors for fatal TAM include preterm delivery before 37 weeks gestation, hyperleucocytosis >100 × 109/L, hydrops foetalis, presence of visceral effusions, severe or progressive hepatic or renal dysfunction and DIC with bleeding29,30,78 (Figure 2). The presence of any of these risk factors or life-threatening symptoms is associated with 1 year survival of 45% and treatment is indicated for this group of children. 35
TAM blasts are exquisitely sensitive to cytarabine, and short courses of low-dose cytarabine can be used successfully to drive blast clearance. Located on chromosome 21, the overexpression of cystathionine-β-synthase, due to gene dosage effect of T21, results in increased intracellular levels of active 1-β-
Treatment of ML-DS consists of multiagent chemotherapy with a backbone consisting of high-dose cytarabine and dose-modified anthracyclines.76,77,84 Survival outcomes in ML-DS are reported to be higher in most studies, compared to children without DS who are diagnosed with AML, with 5-year OS of 80–90%.76,84,85 The general survival advantage is in part due to marked chemosensitivity of ML-DS blasts, leading to development of dedicated reduced-intensity ML-DS treatment protocols. Detection of MRD at the end of induction by either flow cytometry or targeted sequencing of GATA1 is a significant prognostic factor for poorer event-free survival (EFS) and predicting risk of relapse.76,77,86 There is limited evidence to date regarding identification of cytogenetic abnormalities that may be predictive of treatment response. Trisomy 8 has been identified as a risk factor for positive MRD at the end of induction and an independent prognostic factor for relapse.76,85
Increased cellular sensitivity to chemotherapy seen in ML-DS cells is also present in non-leukaemic cells, contributing to increased treatment-related morbidity and mortality in children with DS compared to those without DS.87,88 Children with DS are at higher risk for prolonged acute hospitalisation and long-term treatment-related side effects such as cardiomyopathy.88,89 Major acute complications include risk of infection,87,90 with up to 15% infection-related mortality reported by some groups.90,91 Retrospective data extraction for children treated on the AML-BFM-2004 protocol found 97% of patients developed at least one infectious complication during therapy, with a total of 157 infectious events occurring in 61 patients. 90 Gram-positive bloodstream infections, particularly viridans group Streptococci, and viral respiratory tract infections were the most common infectious complications identified. 90 Infectious deaths were due to viral aetiologies; two patients died due to respiratory syncytial virus pneumonia and one due to herpes simplex virus encephalitis. 90
Recent trials by both the Japanese Paediatric Leukaemia Group and Children’s Oncology Group (COG) have attempted to reduce acute infectious toxicity by reducing treatment intensity in the standard risk group, identified as those with negative MRD at the end of induction.77,84 In the COG AAML1531 study, omission of high-dose cytarabine for the standard risk group resulted in worse outcomes and inferior EFS, suggesting a key role of high-dose cytarabine in the treatment of ML-DS, despite associated toxicities. 77 Determining the appropriate dose intensity of curative chemotherapy while minimising treatment-related toxicity remains a challenge, and reductions in treatment intensity need to be balanced against the risk of jeopardising the high cure rate of ML-DS and poor prognosis of relapsed or refractory (R/R) ML-DS (Table 1).
Summary of key trials in ML-DS. Inclusive of 1992 seminal paper and key updates since 2016.

POG, Pediatric Oncology Group; CNS, central nervous system; AML, acute myeloid leukaemia; EFS, event-free survival; EOI, end of induction; ML-DS, myeloid leukaemia of Down syndrome; MRD, minimal residual disease; HD-AraC, high-dose cytarabine; HR, high-risk; OS, overall survival; SR, standard risk.
In contrast to the favourable outcome of primary ML-DS, the outcome for R/R ML-DS is dismal, with 20–25% OS, compared to 40% OS in R/R non-DS AML.93,94 There are few clinical trials and no standardised treatment for R/R ML-DS. In retrospective reviews of iBFM-AML and Japanese study groups, the median time to relapse was 6.8 and 8.6 months, respectively.93,94 Duration of first remission (>12 months) and attainment of remission after relapse was associated with improved OS.93,94 Salvage chemotherapy, typically a fludarabine, high-dose cytarabine and G-CSF (FLAG-) based regimen, resulted in complete remission in approximately half of the patients with R/R ML-DS.93,94 Treatment with chemotherapy alone resulted in death from progressive disease in the majority of patients, although a small number were able to maintain a durable complete remission with chemotherapy alone.93,94 Consolidation with allogeneic haematopoietic stem cell transplant (HSCT) in ML-DS was associated with better OS compared to chemotherapy alone, but only in those patients who achieved complete morphological remission prior to transplant. 94 In a recent retrospective series by Hitzler et al. 95 transplant-related mortality was 24%, compared to 15% in R/R non-DS AML (hazard ratio 2.52). Risk of relapse post-transplant for R/R ML-DS was significantly higher compared to non-DS AML, with 3-year relapse rates of 62% in patients with ML-DS (versus 37% in non-DS AML) even in those who were in morphological remission prior to HSCT, 95 highlighting a need for better methods of evaluating MRD and new treatments to achieve a deeper remission prior to HSCT. 95
The high incidence of treatment-associated morbidity with conventional chemotherapy and dismal outcomes of R/R disease makes the use of novel or targeted therapies appealing in ML-DS. To date, there is limited evidence for use in this setting as children with ML-DS are typically excluded from current clinical trials of new and targeted therapies. 96
Pre-clinical evaluation of novel agents in patient-derived xenograft (PDX) mouse models of ML-DS has identified several promising therapies. Epigenetic therapies, including the DNA hypomethylating agent azacitidine, histone deacetylase (HDAC) inhibitors such as vorinostat, panobinostat or romidepsin, and the EZH2 inhibitor GSK126, have been used alone or in combination, with anti-leukaemic activity and prolonged survival in PDX models compared to vehicle.97–99
HDAC inhibitors represent an attractive treatment option for ML-DS, as reduced autophagy secondary to activation of the IGF/IGF1R/PI3K/mTOR pathway is a distinguishing feature of ML-DS blasts.37,99 HDAC inhibitors repress autophagy in ML-DS to a critical level, resulting in mitochondrial mass accumulation, production of reactive oxygen species and cell death. 99 HDAC1/2 inhibitors in ML-DS cell lines induced cell cycle arrest and apoptosis in vitro, and resulted in prolonged survival in PDX mouse models. 99 A reported case study of single agent vorinostat in a child with a second relapse of ML-DS after HSCT demonstrated an initial haematological response with clearance of peripheral blasts shortly after commencing vorinostat therapy (230 mg/m2 daily), with subsequent dose reductions required due to grade 3 nausea. 100 Unfortunately, the patient experienced frank disease progression 2 months later. Two children with relapsed ML-DS were enrolled in the T2016-003 Therapeutic Advances in Childhood Leukaemia and Lymphoma (TACL) Phase I study examining the use of decitabine and vorinostat in combination with FLAG chemotherapy in R/R AML.96,101 One patient achieved negative flow MRD following the first and only cycle of FLAG in combination with vorinostat (180 mg/m2 × 5 days) and decitabine (10 mg/m2 × 5 days), whilst the other patient died from progressive disease.96,101 The surviving patient went on to receive a matched unrelated HSCT and remains in complete remission more than 3 years post-transplant. 96 When used in combination, panobinostat and azacitidine were more effective in inducing remission and prolonging survival in PDX models compared to single agent or conventional chemotherapy (cytarabine and daunorubicin). 97 There is a single reported case describing use of azacitidine in a child with a second relapse of ML-DS after transplant who achieved complete remission following two cycles of azacitidine (75 mg/m2 × 7 days). 102 The patient experienced progressive disease following cycle 3 of azacitidine but was salvaged with further cycles and remains in complete remission 8 months after receiving the fifth and final cycle of azacitidine. 102
Despite increasing evidence for use of the Bcl2 inhibitor, venetoclax, in paediatric R/R AML, there has been limited exploration of its use for ML-DS. The Berlin-Frankfurt-Munster (BFM) group report three patients with relapsed ML-DS treated with venetoclax, none of whom achieved complete remission. 94 Barwe et al. 103 examined the use of the combination of azacitidine and the Bcl2 inhibitor venetoclax in PDX mouse models, with prolonged survival seen in combination therapy mice compared to either agent alone. Transcriptome analysis of cells harvested from mice following treatment demonstrated synergistic downregulation of cytokine signalling as the likely mechanism of action for venetoclax combined with azacitidine. 103 Selinexor, a nuclear exportin protein XPO1 inhibitor, has also been tested in ML-DS PDX models alone and in combination with venetoclax. Selinexor monotherapy demonstrated greater efficacy in reducing leukaemic burden and improving survival compared to single agent venetoclax or combination therapy. 104
Pharmacological inhibition of KIT with tyrosine kinase inhibitors has been trialled in mice transplanted with ML-DS cell lines. 55 Proto-oncogene KIT codes for a receptor tyrosine kinase that plays a crucial role in regulating haematopoietic stem cell proliferation and survival, 105 and dysregulation of KIT signalling or gain of function mutations acts as a leukaemic driver in non-DS AML and ML-DS.55,106 Administration of KIT inhibitors imatinib, dasatinib or ripretinib resulted in differentiation of blasts into mature myeloid cells in preleukaemic cell lines (T21/GATA1s) transplanted into mice and reduced engraftment of TAM and ML-DS blasts. 55
The Wee1 kinase inhibitor MK-1775 demonstrated modest activity as a single agent, but when trialled in combination with cytarabine enhanced cytarabine-induced DNA damage and apoptosis in ML-DS cell lines (CMK and CMY) and ex vivo primary ML-DS samples. 107 MYC inhibitor MYCi361 has therapeutic potential in ML-DS by disrupting the RUNX1A/MAX interaction. 44 MAX, a cofactor of MYC, has been proposed to be critical in the synergy between GATA1s and RUNX1A, and inhibition with MYC inhibitor MYCi361 induced apoptosis and partial differentiation in ex vivo and PDX ML-DS samples. 44
While there are promising data on possible targets of future therapies in ML-DS for refractory disease, current treatment of R/R disease is limited to toxic conventional chemotherapy with very poor outcomes. Future strategies for treatment should consider the combination of newer agents with conventional treatment for improved outcomes.
Down syndrome-associated acute lymphoblastic leukaemia
The epidemiology of DS-ALL is strikingly different to that of ML-DS. The incidence of ALL is 10–20 times higher in children with DS compared to children without DS 25 and rarely occurs in infants.108,109 DS-ALL is almost exclusively B-cell precursor immunophenotype, as opposed to T-cell immunophenotype ALL. 108 Historically, the prognosis of DS-ALL was relatively poor compared to children without DS, due to a higher risk of induction failure, relapse, treatment-related toxicities and infection.109,110 However, recent data indicates that outcomes for children with DS-ALL are improving with risk-adapted, personalised therapies.118,121,125
Molecular drivers in DS-ALL
DS-ALL is not a single biological entity, but rather displays variable genetic heterogeneity. 67 The common cytogenetic changes seen in childhood ALL without DS are less common in children with DS. Patients with DS-ALL have a decreased incidence of favourable cytogenetic features, such as the ETV6-RUNX1 translocation and high hyperdiploidy, compared to children without DS. 109 Interestingly, children with DS-ALL also have a decreased incidence of unfavourable chromosomal aberrations, such as BCR-ABL and MLL-AF4,67,109 and are more likely to present with somatic constitutional T21 alone without additional cytogenetic changes (40.3% in DS-ALL compared to 6.9% in non-DS-ALL in one cohort study). 109 IKZF1 deletions have been found more frequently in DS-ALL compared to non-DS-ALL. 111 DS-ALL is also more likely to present with CRLF2 rearrangements and JAK2 mutations, occurring in up to two-thirds of cases.112,113 In a recent series by Li et al., 113 50% of patients with CRLF2-rearranged DS-ALL demonstrated co-occurrence of JAK2 mutations, whereas JAK2 mutations were absent in patients without CRLF2 rearrangement. 113
Activating mutations in JAK2, NRAS and KRAS, and CRLF2 rearrangement are key drivers in development of DS-ALL. 114 The most common drivers for the development of DS-ALL are activating mutations in inter- or intra-chromosomal rearrangement of the CRLF2 gene. This results in overexpression of CRLF2, with subsequent increased lymphopoiesis of the immature B-lineage immunophenotype, observed in up to 60% of DS-ALL cases.113–115 Activating KRAS mutations cooperate with T21 to block B-cell differentiation and promote cell proliferation and self-renewal capacity. 116 Additional recently implicated cooperating abnormalities include those affecting genes such as HMGN1, DYRK1A, IKZF1 and PAX5, 114 with one DS-ALL mouse model requiring mutations in JAK2, overexpression of CRLF2, PAX5 deficiency, IKZF1 loss and T21 to induce B-ALL in vivo. 117 HMGN1 overexpression leads to global suppression of H3K27 trimethylation (H3K27me3), which in turn results in upregulation of genes that drive phenotypic changes, including leukaemogenesis in DS-ALL. 117
Treatment of DS-ALL
Current treatment approaches for DS-ALL align with those of children without DS, with treatment on prospective clinical trials, stratified by MRD response. Treatment consists of multiagent chemotherapy, central nervous system-directed therapy and the use of immunotherapy in some patients. Treatment is tailored for DS patients by most international trial groups through limiting intravenous methotrexate dosing, additional folinic acid rescue after intrathecal methotrexate and limiting anthracycline exposure. 118
Children with DS-ALL are often excluded from Phase I and II clinical trials based on the increased risk of serious adverse events and life-threatening complications in children with DS. 119 The 5-year EFS and OS in children with DS-ALL is significantly inferior to children without DS (79.2% and 86.8%, respectively, versus 87.5% and 93.6% in patients without DS; p < 0.0001), 118 particularly in children older than 6-10 years of age.109,118 This is further increased in those with high risk cytogenetics, with relapse risk in IKZF1-deleted DS-ALL significantly higher than matched non-DS controls,111,120,122 quoted at 37.1% versus 13.2% at 5 years in a large international cohort study. 110 CRLF2 and/or JAK mutations did not confer adverse outcomes in DS-ALL in pooled analysis of 317 patients treated on COG studies, 123 however a larger COG analysis (743 DS-ALL and 20,067 non-DS ALL patients) demonstrated a differential prognostic impact of CRLF2 overexpression according to DS-ALL risk group. 118 In HR DS-ALL, CRLF2 overexpression compared to CRLF2-normal was associated with inferior EFS, while there was no negative prognostic impact for SR DS-ALL patients. 118 Cumulative risk of ALL relapse is higher in patients with DS compared to those without DS (11.5% versus 9.1% 5-year cumulative incidence of relapse), 118 and outcomes following relapse are dismal, with 5-year OS following relapse less than 20%. 118
Similar to ML-DS, children with DS-ALL are more susceptible to treatment-related toxicities. 124 Children with DS-ALL experience significantly higher rates of mucositis, infections, hyperglycaemia, non-CNS thrombosis and seizures compared to non DS-ALL patients treated on the same protocol,118,125 and over the course of treatment experience longer inpatient admissions. 126 Often overlooked, DS-associated comorbidities such as atlantoaxial instability, structurally challenging airways, inhalational anaesthetic-induced bradycardia and post-extubation stridor affect anaesthetic safety for children during leukaemia treatment. 127
Patients with DS are particularly sensitive to the anti-folate agent methotrexate, possibly due to altered pharmacokinetics or gene dosage associated with T21, as the gene for the transmembrane protein responsible for transporting methotrexate intracellularly (SLC19A1) is located on chromosome 21, 124 and gene dosage-related overexpression may result in greater uptake into tissues. Current practice in most cooperative groups is to limit high-dose methotrexate by using either a capped dose or a dose escalation strategy based on individual tolerance.126,128 Folinic acid rescue is an important supportive care measure to minimise methotrexate-associated toxicity. Due to the limited number of DS patients in ALL clinical trials, it is difficult to determine if DS patients are at increased risk of other therapy-related complications, such as asparaginase-associated toxicity.
Infection-related morbidity and mortality remain a significant risk in children receiving therapy for DS-ALL. Comorbidities associated with DS, including immunodeficiency (specifically, defects in switched memory B-cells and mild T-cell dysfunction), cardiac defects, airway anomalies and pulmonary dysfunction, contribute to the increased susceptibility of patients with DS-ALL to infectious complications.126,129,130 Children with DS-ALL experience a higher risk of death during and post-induction, primarily due to infection. 109 A recent review of four COG trials reported an incidence of death in induction at 3.4% in children with DS and ALL (versus 0.8% in children with ALL and without DS) and 5-year cumulative incidence of death in remission at 4.9% (1.7% in children with ALL and without DS). 118 Infectious complications are more frequent in all phases of therapy, including during maintenance treatment. 118 Children with DS-ALL are more likely to develop grade 3–4 cellulitis than children with non-DS-ALL, suggesting a need for close vigilance for cutaneous infections and skin hygiene care. 131
In an attempt to address these issues for children with DS-ALL, cooperative group clinical trials have focused on standardising dose modifications and providing guidance around intensified supportive care regimens for children with DS-ALL. 67 This guidance includes a strong recommendation for consideration of antifungal and antibiotic prophylaxis, particularly in intensive treatment phases, monitoring and prompt treatment of hypogammaglobulinaemia to reduce risk of infection, annual influenza vaccination and an acknowledgement that most clinical trials groups avoid cranial radiation in children with DS. 67 Implementation of DS-specific supportive care modifications, in addition to reduction of anthracycline dosing in the recent COG AALL1131 trial successfully reduced induction mortality in HR DS-ALL patients when compared to previous HR DS-ALL cohorts. 132 Despite this, induction death rate remained significantly higher than in non-DS HR ALL patients (3.6% versus 1.7%, p = 0.035), suggesting supportive care strategies alone are not adequate to reduce disparity in treatment-related mortality seen in DS-ALL and that intensified conventional therapy remains a challenge, particularly for HR DS-ALL patients. 132
There is potential that clinician-driven modifications to treatment protocols for children with DS-ALL due to the possible risk or presence of toxicities may also contribute to their inferior outcomes. This is a key paradox in the treatment of DS-ALL, where treatment-related toxicities limit the ability to intensify treatment. A study from the Nordic Society of Paediatric Haematology and Oncology indicated that clinicians were less willing to increase the maintenance dose of oral methotrexate and 6-mercaptopurine for children with DS-ALL due to concerns regarding toxicity, ultimately dosing children with DS-ALL up to 25% less. 133
It has also been reported that up to 40% of patients with DS have received chemotherapeutic dose reductions during treatment, especially during high-dose methotrexate blocks in an attempt to reduce therapy-related toxicity, 134 leading to suboptimal treatment for children with DS-ALL and an increased risk of relapse.
Further analysis of acute toxicities, including infection, neurotoxicity, venous thrombosis, pancreatitis and bone toxicity, in children with DS-ALL is required (Figure 3). An in depth and up to date analysis of key toxicities may allow for adaptive treatment regimens tailored to the increased vulnerability of patients with DS without compromising on cure. 126

Future research directions in DS-leukaemia.
In an attempt to increase leukaemia-free survival and minimise acute toxicity from conventional chemotherapeutics, immunotherapeutic agents such as blinatumomab have been incorporated into the backbone of the upfront COG (NCT03914625), Associazione Italiana di Ematologia e Oncologia Pediatrica (AIEOP)/BFM (NCT03643276) and European ALLTogether (NCT03911128) clinical trials. The COG AALL1731 study observed a higher incidence of blinatumomab-associated seizures in patients with DS versus non-DS-ALL (17% versus 4% in historical cohort), although this increased frequency was only observed in children older than 10 years.135,136 All patients recovered fully with no sequelae and the majority were able to resume blinatumomab without further seizures. 135 Anti-epileptic prophylaxis is now strongly recommended by COG and AIEOP/BFM study groups for patients with DS-ALL receiving blinatumomab. Another immunotherapy agent, inotuzumab ozogamicin (InO) (CD22-targeted antibody-drug conjugate), has been trialled as a single agent salvage therapy in relapsed ALL in patients with DS. In an international retrospective review of patients who received InO in a compassionate use program, three of four patients with relapsed ALL and DS achieved MRD-negative remission following InO therapy. 137 A recent COG trial AALL1621 investigated InO in R/R B-cell ALL, including for three patients with DS-ALL after first relapse (out of total 48 patients), with two patients achieving complete remission. 138 Of these, one patient proceeded to consolidative HSCT and remains in remission after 18 months of follow-up; the other received InO for 7 months but relapsed and died 16 months after commencing treatment. 138
In addition, children treated for HR DS-ALL who are MRD positive at the end of consolidation are eligible to enrol on a COG Phase II clinical trial investigating tisagenlecleucel, an autologous chimeric antigen receptor (CAR) T-cell therapy (NCT03876769). A recent post hoc analysis of three clinical studies identified 16 patients with R/R DS-ALL who were treated with tisagenlecleucel. 139 Outcomes demonstrated comparable efficacy and safety outcomes to children without DS, without treatment modification. The remission rate was 88% within 3 months of infusion, and the probability of remaining in remission at 1- and 3-year post-infusion was 57% and 38%, respectively (Table 2). 139 CAR T-cell-related toxicity in children with DS-ALL was comparable to children with non-DS-ALL, including the course and management of cytokine release syndrome and infective complications, 139 making novel immunotherapy agents an attractive therapeutic option for children with DS-ALL.
Summary of key trials in DS-ALL since 2016.
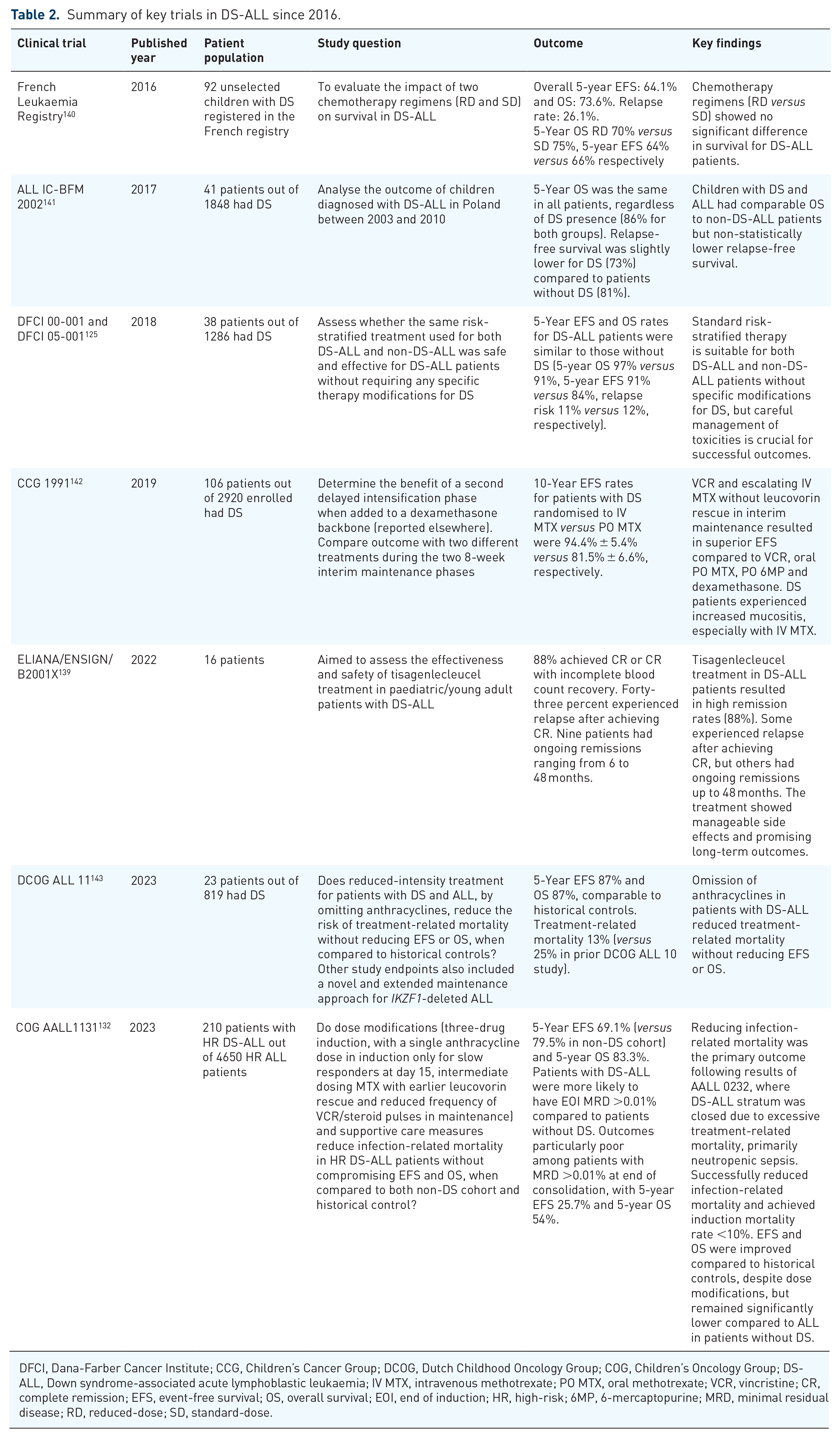
DFCI, Dana-Farber Cancer Institute; CCG, Children’s Cancer Group; DCOG, Dutch Childhood Oncology Group; COG, Children’s Oncology Group; DS-ALL, Down syndrome-associated acute lymphoblastic leukaemia; IV MTX, intravenous methotrexate; PO MTX, oral methotrexate; VCR, vincristine; CR, complete remission; EFS, event-free survival; OS, overall survival; EOI, end of induction; HR, high-risk; 6MP, 6-mercaptopurine; MRD, minimal residual disease; RD, reduced-dose; SD, standard-dose.
The role of HSCT in DS-ALL is unclear, and large prospective trials are lacking. Early series reported transplant-related mortality up to 75%, exceeding leukaemia relapse as primary cause of post-transplant mortality.144,145 Respiratory toxicities, including pulmonary haemorrhage, pneumonitis and upper airway obstruction secondary to mucositis, accounted for a significant proportion of toxicity-related morbidity and mortality, with reported incidence of fatal pulmonary toxicity as high as 26%.144,145 In more recent series, transplant-related mortality ranged from 18% to 25%.146,147 Three-year disease-free survival was 24%, with approximately half of patients experiencing relapse post-transplant. 146 In childhood ALL more broadly, improved supportive care following HSCT has led to reduced TRM over time; however, relapse rates have remained static. 148 In line with this observation and in contrast with earlier series, leukaemic relapse is now the most common cause of post-transplant mortality in children treated for DS-ALL.146,147 Relapsed disease was the most common indication for transplantation, with the majority of patients undergoing HSCT in CR2 or greater.146,147 Myeloablative conditioning regimens, similar to those used in non-DS-ALL, were successfully used without excessive early toxicity. 147 The high burden of relapse post-transplant suggests better leukaemia control prior to HSCT, whilst avoiding excessive toxicity of standard ALL chemotherapy, which needs to be addressed to improve outcomes of HSCT in DS-ALL.
High rates of treatment-related morbidity in upfront and relapsed treatment and dismal outcomes of R/R DS-ALL have seen an increasing interest in the use of novel targeted therapies for children with DS-ALL. JAK inhibitors such as ruxolitinib added to conventional chemotherapeutic regimens are currently undergoing Phase II clinical trials in paediatric CRLF2-rearranged Philadelphia chromosome-like ALL in patients without DS and may provide options for future clinical investigation in patients with DS.149,150 Bagashev et al. 151 examined the use of thymic stromal lymphopoietin receptor (TSLPR)-redirected CAR T-cell in CRLF2-rearranged DS-ALL PDX models and demonstrated potent inhibition of leukaemic proliferation in vivo. Delayed co-administration of ruxolitinib 14 days after infusion of TSLPR-redirected CAR T-cell resulted in complete peripheral clearance of leukaemic blasts, 151 suggesting a potential role for maintenance-style inhibitor therapy following CAR T-cell therapy in DS-ALL. The role of RAS/MAPK pathway activation in cell proliferation and self-renewal capabilities in DS-ALL make MEK inhibitors an attractive therapeutic option. MEK inhibitors, including MEK1/2 inhibitor trametinib, inhibit essential downstream mediators of the RAS/MAPK pathway. Therapy with trametinib alone or in combination with vincristine significantly reduced leukaemic burden in DS-ALL PDX mouse models. 116
Late effects in survivors of DS-leukaemia
There is limited data regarding long-term sequelae of cancer therapy in adults with DS who survive childhood leukaemia. It is known that people with DS, without a diagnosis of leukaemia, experience a high burden of chronic health conditions in older age, however, the additional toxicities associated with treatment for DS-leukaemias may increase this burden. 152 Compared to survivors of childhood leukaemia without DS and adults with DS without a history of leukaemia, the risk of late mortality is higher in DS-leukaemia survivors,153,154 primarily due to late relapse. 154 In one study comparing 154 survivors of DS-leukaemia to 581 survivors of leukaemia without DS, 83% of DS patients had developed at least one chronic health condition. 89 These patients, when matched for leukaemia subtype and treatment exposure, were more likely to experience severe, life-threatening or fatal late chronic conditions, in particular endocrinopathies, hearing loss and cataracts. 89 Survivors of DS-associated leukaemia were also more likely to experience poorer health-related quality of life compared to survivors without DS.89,155 While the authors recognise that it is difficult to compare DS-leukaemia survivors with adults survivors of childhood leukaemia without DS, it is considered that survivors may be more prone to, or have acceleration of, chronic conditions in DS. 89 Interestingly, secondary malignant neoplasms are rare in adult survivors with DS compared to those without DS. 89
Adult survivors of both ML-DS and DS-ALL have poorer neurocognitive outcomes and functioning compared to adults with DS and no history of leukaemia. 156 Adult survivors treated for DS-ALL experience deficits across the entire neurocognitive range and poorer adaptive function compared to adult survivors of ML-DS. 156 The presence of physical and intellectual disabilities in DS may also impact on reporting of symptoms, presentation and screening for complications and effective promotion and implementation of positive health behaviours, such as engaging in regular physical activity. 127
Particularly concerning is the increased risk of anthracycline-induced cardiomyopathy seen in adults with DS who survive childhood leukaemia,88,92,157 including patients treated with reduced-intensity DS-specific protocols. In one trial (POG AML 9421), the incidence of cardiomyopathy was 17.5% in 57 patients, with three patients dying of congestive heart failure. 88 Potentially impacting the increased cardiac toxicity seen in patients with DS is the increased incidence of underlying congenital heart disease (CHD), with 24 patients (42%) on the POG AML 9421 trial having documented CHD and that 50% of those who developed cardiomyopathy following treatment had a background of CHD. 88
The pathogenesis of anthracycline-related cardiotoxicity has been linked to intracardiac synthesis of alcohol metabolites by carbonyl reductase 1 (CBR1), located in the DSCR (21q22.12). 158 Cardiac tissue in individuals with DS displays higher CBR1 mRNA levels and activity compared to individuals without DS, which may contribute to the increased risk of anthracycline-related cardiotoxicity in patients with DS. 158 The threshold for safe anthracycline exposure in children with DS remains unknown. In a single retrospective cohort study, there was no significant relationship between CHD in patients with DS and risk of cardiomyopathy, but a higher incidence of cardiomyopathy-related deaths was observed in those with CHD, which may be due to lower cardiac reserve at baseline and reduced ability to tolerate a decrease in cardiac function. 88 Adults with DS have a high risk of cardiometabolic comorbidities, including diabetes mellitus, hyperlipidaemia and obesity, and inequitable access to preventative health care globally, compounding the cardiovascular late effects of chemotherapy.127,159 Close monitoring of cardiac function remains a crucial component of long-term care, although currently, there are no dedicated evidence-based guidelines regarding cardiac follow-up for adult survivors of DS-leukaemia.
Understanding the long-term impacts of leukaemia therapy is paramount, as both survival for children with DS diagnosed with leukaemia and the overall life expectancy of adults with DS continue to increase. Further research to determine screening modalities and intervals to improve quality of life and health outcomes in survivors of DS-leukaemia is critical (Figure 3).
Conclusion
Over the last decade, there has been improved understanding of T21-driven leukaemias and their associated molecular and epigenetic drivers. Models for leukaemogenesis provide greater knowledge of the complex interaction between T21 and additional acquired genetic mutations that drive the transformation of TAM to ML-DS and the development of leukaemia in DS-ALL. Such insight can be applied to identify novel therapeutic targets for patients with preleukaemia and leukaemia in DS, as well as non-DS patients with HR leukaemia and somatic chromosome 21 aneuploidy or iAMP21.
Our review highlights areas for future research that are essential to improve outcomes for children with DS treated for either ML-DS or DS-ALL (Figure 3). For patients with TAM and ML-DS, the emergence of GATA1 PCR monitoring and detection of MRD are promising techniques to determine HR patients and to identify patients with TAM who may progress to ML-DS and may in turn benefit from additional therapy. Optimisation of pre-clinical models that truly reflect the spectrum of TAM to ML-DS progression will assist efforts to explore prevention of ML-DS in patients in future. These models may also serve to further understand the stem cell leukaemia population that may persist in driving relapse in DS-leukaemias.
Furthermore, inclusion of DS patients in standardised treatment protocols for ML-DS and DS-ALL and use of specific risk stratification tools for DS patients may allow for improved outcomes without associated toxicity. Management of relapsed/refractory DS-associated leukaemias remains a challenge, and international collaboration is vital to assess the effectiveness of emerging targeted therapies in monotherapy and as combination regimens. Emerging novel technologies such as CRISPR-Cas9 may assist with determining synthetic lethal combinations for specific use in DS-associated leukaemias.
Limiting treatment-related toxicities for children treated for DS-associated leukaemias remains a challenge, with a fine balance between improving survival and reducing risk of relapse, and the increased burden of chronic health conditions later in life. Biomarker discovery using next generation sequencing data or genome-wide association studies may identify additional risk factors that can predict development of treatment-related toxicities in DS-leukaemias. An understanding of the unique complications that DS patients face in treatment, tailored monitoring for complications of therapy and engagement in long-term follow-up services may help to reduce the severity of late complications in DS-leukaemia survivors. Expanding the use of immunological and targeted therapies in both ML-DS and DS-ALL provides an exciting opportunity to limit toxicities and improve long-term survival.