
Abstract
In the mythology of Ancient Greece, there was often a creative tension between the opposing forces of the gods Apollo and Dionysius, the two sons of Zeus. The Apollonian force was considered to be rational and lifegiving, whilst Dionysian forces were chaotic and elemental. Acute myeloid leukaemia is characterised by the clash of these forces: the chaotic proliferation of immature myeloid cells in the bone marrow overcomes the normal, orderly production of healthy blood cells. DNMT3A mutations occur early in the leukaemogenic process and may even act as “founder” mutations – the first step in a pathway towards malignant transformation. As such, these mutations may represent a Dionysian agent of disorder, inciting the chaotic myeloid proliferation and arrest of differentiation which are hallmarks of AML. This review will focus on the role of DNMT3A mutations in leukaemia pathogenesis, their influence on prognosis, and the potential for therapeutic targeting.
Introduction
In the mythology of Ancient Greece, there was often a creative tension between the opposing forces of the gods Apollo and Dionysius, the two sons of Zeus. The Apollonian force was considered to be rational and lifegiving, whilst Dionysian forces were chaotic and elemental. Acute myeloid leukaemia (AML) is characterised by the clash of these forces: the chaotic proliferation of immature myeloid cells in the bone marrow overcomes the normal, orderly production of healthy blood cells [O’Donnell et al. 2012; Estey, 2013; Ferrara and Schiffer, 2013]. AML is a malignant disorder of myeloid cells resulting in symptoms such as fatigue, susceptibility to infection, and easy bruising, and is the most common leukaemia in adults [O’Donnell et al. 2012; Ferrara and Schiffer, 2013]. Highly variable, AML is likely a composite of several disease entities, each displaying a subtly distinct clinical profile and a discrete genetic background. Current treatment options include chemotherapy and haemopoietic stem cell transplants (HSCT), with 5-year survival hovering at around 25% [Ferrara and Schiffer, 2013].
In the last few decades the importance of genetic abnormalities in the initiation and progression of malignant disease has become abundantly clear [Grimwade et al. 1998, 2001]. AML is risk stratified according to chromosomal anomalies into good, intermediate, and poor risk categories, which help inform prognosis [Grimwade et al. 1998, 2001]. Specific chromosomal anomalies even offer therapeutic targets: the prognosis of acute promyelocytic leukaemia, which commonly features t(15;17), has been improved to over 80% survival at 5 years due to the development of all-trans retinoic acid (ATRA) treatment [Grimwade et al. 2014]. Not all leukaemia genomes are so obliging, however. Nearly 50% of AML cases are categorized as cytogenetically normal (CN-AML) and thus by default fall into the ‘intermediate’ risk category, despite considerable variation in disease behaviour and outcome [Vardiman et al. 2009; Lin and Smith, 2011; Estey, 2013]. Advances in genome sequencing have highlighted molecular genetic anomalies which are frequently found in AML that has no detectable chromosomal abnormalities on karyotyping or fluorescence in-situ hybridization (FISH) analysis [Estey, 2013]. A number of these mutations, in genes such as NPM1, FLT3, and CEBPA, have been incorporated into the European LeukemiaNet prognostic stratification model [Döhner et al. 2010], although there remain myriad other mutations with uncertain prognostic significance [Patel et al. 2012].
Awareness of the complexity of the leukaemic genome has been further heightened by the discovery of mutations in genes bearing epigenetic functions. Genes concerned with DNA methylation, such as DNMT3A, TET2 and IDH1 and IDH2, and those concerned with post-translational histone modifications, such as ASXL1 and EZH2, have been reported to occur in a significant proportion of AML [Shih et al. 2012]. DNMT3A, in particular, is mutated in 15–25% of AML and is thought to be a ‘founder’ mutation: present in the early preleukaemic stem cells [Shlush et al. 2014]. As the potential initiating genetic lesion, DNMT3A mutations may represent the Dionysian agent of disorder, inciting the chaotic myeloid proliferation and arrest of differentiation which define AML. This review will focus on the role of DNMT3A mutations in leukaemia pathogenesis, their influence on prognosis, and the potential for therapeutic targeting.
DNA methylation in leukaemia
Epigenetic modifications are heritable changes in gene expression that are not a result of changes to coding DNA. DNA methylation is one of the most important epigenetic methods of controlling gene expression, occurring solely at the site of CpG residues in eukaryotic cells [Suzuki and Bird, 2008; Shih et al. 2012]. CpG residues (cytosine molecules with an adjacent guanine) are not evenly distributed through the genome but instead occur in clusters, or ‘CpG islands’ [Suzuki and Bird, 2008; Shih et al. 2012]. Oceanographers estimate that humans have explored no more than 5% of the world’s oceans. Considering the complex and poorly understood nature of epigenetic processes, it is perhaps appropriate that the position of CpG residues in the genome is described through a marine analogy. In addition to CpG islands, CpG residues may occur on island ‘shores’, defined as less than 2 kb from an island, and ‘shelves’, which are 2–4 kb from an island. CpG residues which are dotted throughout the genome are considered to be in ‘open sea’ regions. Approximately 60% of human genes have CpG islands in the gene promoter region, with approximately 13,000 regions constitutively unmethylated [Suzuki and Bird, 2008; Oki and Issa, 2010]. Methylation of CpG islands stably represses gene expression via recruitment of corepressor complexes [Takahashi, 2011]. During development a small number of these promoter regions are methylated, contributing to important functions such as X-inactivation and genomic imprinting [Gutierrez and Romero-Oliva, 2013].
Global methylation patterning (methylation of intron, exon and transposon sequences) plays a role in gene expression control and maintenance of genomic stability [Saied et al. 2012]. While most crucial during embryonic patterning and development, DNA methylation remains dynamic throughout an organism’s life. For example, as haemopoetic stem cells (HSCs) differentiate, genes concerned with maintenance of stem cell status are repressed, while genes involved with differentiation in specific cell lineages are expressed [Borgel et al. 2010]. CpG promoter region methylation is generally considered to be irreversible past the embryonic stage, persisting through mitosis, while nonpromoter region methylation can be reversed [Oki and Issa, 2010]. Reversal of non-CpG island methylation can come about through hydroxylation of methylated cytosine residues. This is carried out by the TET family of enzymes and appears to counter the inhibitory action of DNA methylation, perhaps acting as a step in a pathway to demethylation [Abdel-Wahab et al. 2009; Kinney and Pradhan, 2013].
DNA methylation is altered in both solid cancers and leukaemia, and can affect cells in three main ways: hypomethylation, hypermethylation, and loss of imprinting. Global hypomethylation has frequently been reported in cancer cells, and it is postulated that this promotes transcription of oncogenes and genes concerned with cell replication [Ley et al. 2010]. Local CpG promoter region hypermethylation, particularly in the promoter regions of tumour suppressor genes, also appears to play an important role in AML pathogenesis [Ley et al. 2010]. A recurring combination of aberrantly hypermethylated genes has been reported as occurring in most subtypes of AML, including genes concerned with differentiation, apoptosis and tumour suppression [Bullinger et al. 2010; Figueroa et al. 2010; Akalin et al. 2012]. Further studies of DNA methylation in AML have revealed a series of subgroups with specific methylation signatures. A total of 11 of the 16 subgroups identified by Figueroa and colleagues were enriched with particular mutations [Figueroa et al. 2010], and the finding of specific methylation signatures associated with genetic lesions such as t(8;21), inv(16), t(15;17) and 11q23 has been observed in a number of studies [Bullinger et al. 2010; Akalin et al. 2012].
DNMT3A mutations
DNMT3A mutations were first reported in AML in 2009. DNMT3A belongs to the family of DNA methyltransferase enzymes, and along with its cousin DNMT3B, is concerned with de novo methylation of CpG islands (see Figure 1). This de novo methylation is crucial for embryonic development, and DNMT3A knockout mice die within 4 weeks of birth [Subramaniam et al. 2014]. DNMT1 plays a role in maintenance of methylation patterns during cell division, while DNMT2 may methylate RNA [Goll et al. 2006; Chédin, 2011]. Finally, DNMTL does not itself methylate DNA but forms heterotetramers with DNMT3A and is thought to potentiate enzymatic methylation activity and specificity [Bourc’his et al. 2001]. DNMT3A mutations, as mentioned above, occur in 15–25% of AML cases but are more prevalent in CN-AML (29–34%) [Ley et al. 2010; Yan et al. 2011; Patel et al. 2012; Shih et al. 2012]. They are also associated with other mutations, such as NPM1, FLT3, IDH1 and IDH2. Patients with DNMT3A mutations are typically older than average, have a higher white cell count, and are more likely to have monocytic or myelomonocytic leukaemia (FAB M4/M5) [Patel et al. 2012]. Interestingly, most studies have found that expression levels of DNMT3A are generally higher in bone marrow samples from AML patients although one study has shown lower expression levels in M4/M5 AML, which is more commonly associated with loss-of-function mutations [Huang et al. 2013]. Moreover, although DNMT3A mutations have not been detected in solid tumours, allelic loss of expression has been noted. These findings indicate that DNMT3A expression may have significance for other types of malignancy besides AML.
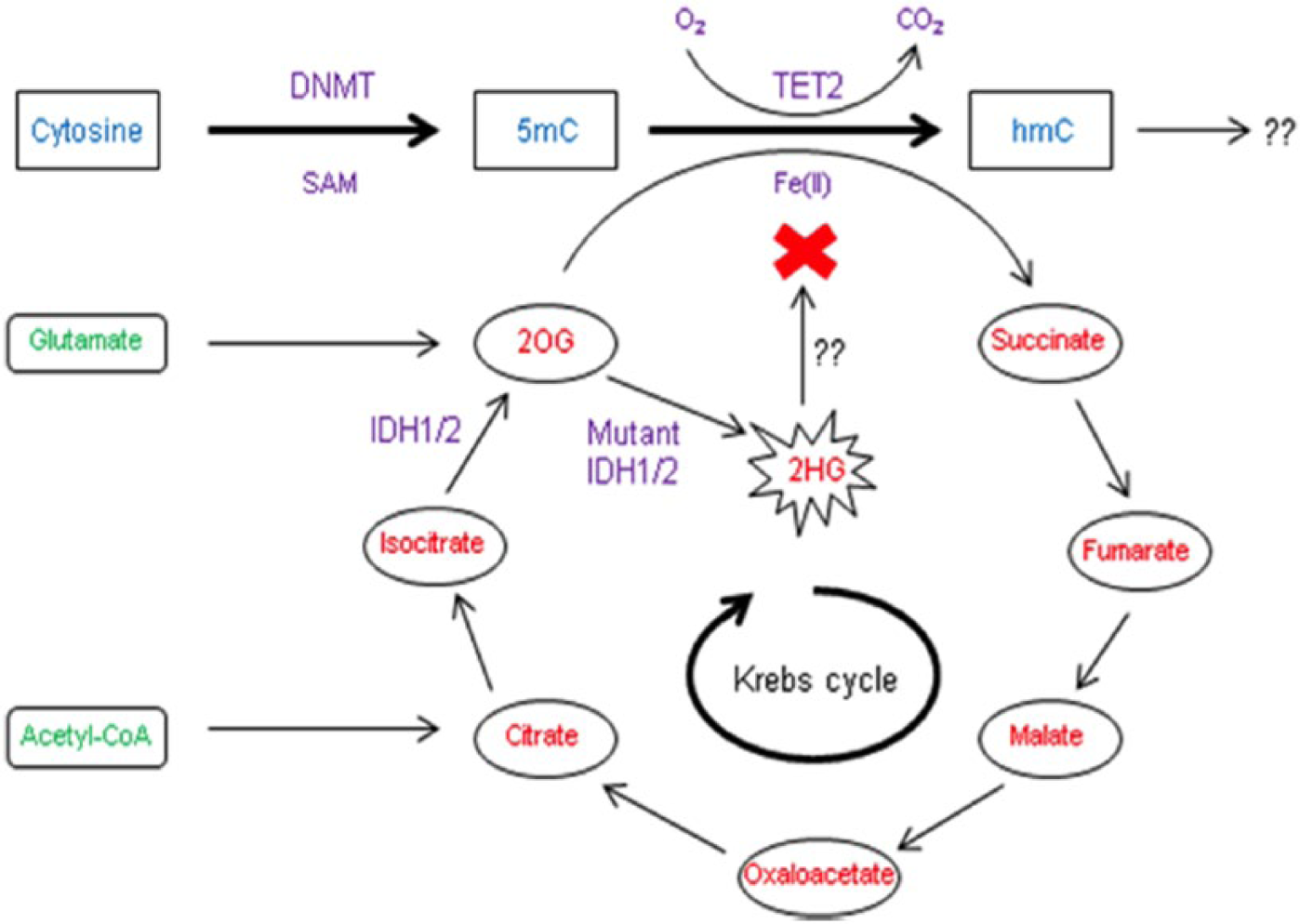
DNA methyltransferase (DNMT) catalyses the addition of a methyl group to the 5-carbon position of cytosine from S-adenosylmethionine (SAM). TET2 and IDH1 and IDH2 are also frequently mutated in acute myeloid leukaemia and influence the hydroxylation of methylated cytosine residues. 5mC, 5-methylcytosine; hmC, 5-hydroxymethylcytosine; TET2, Tet methylcytosine dioxygenase 2; 2OG, 2-oxoglutarate; 2HG, 2-hydroxyglutarate; IDH, isocitrate dehydrogenase.
Mutations affecting DNMT3A may result in missense, frame-shift and nonsense alterations. Approximately 60% of DNMT3A mutations are missense mutations occurring at the R882 codon, which is in the enzyme’s catalytic domain [Holz-Schietinger et al. 2012]. R882H is the most commonly occurring mutation, resulting in arginine-to-histidine change, and like other DNMT3A mutations, is typically heterozygous. Continued expression of both mutated and wild-type DNMT3A in heterozygous cells, observed in vitro, suggests a dominant negative or possibly a neomorphic gain of function role for DNMT3A mutations in AML. Recurring single-site mutations imply alteration of enzyme function, which offers mutated cells a selective advantage, in contrast to typical loss-of-function mutations, which tend to arise at various points within a gene [Ley et al. 2010]. Holz-Schietinger and colleagues found that mutations at the R882 site affect DNMT3A oligomerization resulting in altered catalytic activity and with possible implications for genomic localization [Holz-Schietinger et al. 2012]. The authors propose that DNMT3A function may also be altered by variations in interactions with binding partners, possibly including regulatory RNA molecules. In addition, demethylating agents such as decitabine may improve survival in DNMT3A mutated AML [Metzeler et al. 2012]. Decitabine reduces methylation: its apparent benefit in DNMT3A mutated AML implies that such mutations do not reduce methylating activity but instead may alter methylation patterns. Furthermore, global methylation levels have been found to be normal in DNMT3A mutated AML [Challen et al. 2012]. This evidence indicates that DNMT3A mutations may affect leukaemogenesis not by loss of function but by some other mechanism.
These data are in contrast to in vitro evidence that suggests R882 mutations may result in a loss of DNMT3A function. Murine R878H (the homologue of human R882H) mutated DNMT3A has been shown to have reduced enzymatic activity and reduced ability to bind to S-adenosylmethionine (SAM), the methyl group donor (see Figure 1). In addition, R878H mutated DNMT3A apparently impairs the methylating function of wild-type DNMT3A [Kim et al. 2013]. Yan and colleagues found that R882H mutations reduced methylation activity twofold [Yan et al. 2011]. Similarly, Russler-Germain and colleagues found that when co-expressed, R882 mutated DNMT3A profoundly impaired the enzymatic function of wild-type DNMT3A, at least in part via inhibition of tetramerization [Russler-Germain et al. 2014]. This evidence indicates that DNMT3A mutations may in fact impair enzymatic function, and that a dominant negative effect may be the more likely explanation. Alternatively, there may be a derailment of the normal function together with the propagation of aberrant function. There are a number of challenges to exactly identifying the in vivo consequences of DNMT3A mutations. First, the precise mechanisms controlling wild-type DNMT3A function and targeting of specific CpG residues are unclear, abrogating confident identification of abnormalities in this pathway. Second, in vitro studies are unlikely to fully replicate the actions of mutated DNMT3A in an organism; for example, when mutated and wild-type DNMT3A oligomers are mixed when already formed, they are stable and interact very little. Conversely, when both mutated and wild-type DNMT3A are expressed by a cell, the mutated protein interacts with the wild-type protein and enzymatic function is reduced [Holz-Schietinger et al. 2012]. Similarly, the consequences of DNMT3A mutations in cultured cells may be quite disparate from those in a complete organism. Different DNMT3A mutations may also have distinct effects within the cell. Overall, there is a significant body of evidence to support the theory that DNMT3A mutations lead to loss of function. However, it is likely that these mutations have additional implications, perhaps through interactions with binding partners or as a result of downstream effects, which may explain the observed changes in methylation profile and response to decitabine [Holz-Schietinger et al. 2012]. Further research is required to elucidate more clearly the consequences of DNMT3A mutations on enzymatic function and methylation patterns.
DNMT3A mutations and their effects on DNA methylation
The precise role of DNMT3A mutations on methylation in leukaemia has not yet been clearly elucidated. DNMT3A-null HSCs show accumulation of phenotypically normal cells but a block in differentiation in a murine model [Challen et al. 2012]. Challen and colleagues performed sequential transplants and not only observed global hypomethylation in the progeny of the DNMT3A null cells, but also identified hypomethylation of the promoter regions of HSC multipotency genes and also genes that have previously been implicated in malignancy. Downregulation of genes concerned with differentiation was also noted. These findings go some way towards explaining the expansion of the HSC compartment in conjunction with reduced differentiation into separate cell lineages [Challen et al. 2012]. Interestingly, Challen and colleagues noted no overall change in the level of methylcytosine in DNMT3A null cells, with relatively low numbers of loci demonstrating differential hypomethylation or hypermethylation [Challen et al. 2012]. It is probable, however, that while the absence of DNMT3A has relatively few direct effects, it has a far larger repertoire of indirect effects. This is suggested by the fact that RUNX1, a direct target of DNMT3A in HSC, has over 1000 binding sites throughout the genome [Challen et al. 2012]. The phenotype of DNMT3A null cells may be dictated not only by direct effects on critical genes but also by global changes in methylation, alterations to histone modifications, and indirect changes in gene expression. However, as mentioned above, DNMT3A null HSC may not be a good model for human DNMT3A mutations in AML as the exact in vivo effect of such mutations has not yet been delineated.
In vivo, DNMT3A mutations do not markedly alter global methylation levels [Ley et al. 2010; Shih et al. 2012]. However, DNMT3A mutated AML does demonstrate differential methylation patterns in certain regions of the genome. Qu and colleagues, on analysis of CN-AML samples, found that cases with DNMT3A mutations showed differential methylation patterns to wild-type CN-AML, with almost all of the altered sites hypomethylated [Qu et al. 2014]. The only family of genes that showed statistically significant differential methylation was the HOX family of genes, encoding proteins such as HOXA5, HOXA9 and PAX [Qu et al. 2014]. This finding has been corroborated by a number of studies showing altered expression of HOX genes in DNMT3A mutated AML [Yan et al. 2011; Ribeiro et al. 2012; Jeong et al. 2014]. HOX genes are critical to normal embryogenesis and hematopoiesis, and have been implicated in leukaemogenesis [Eklund, 2011; Yan et al. 2011; Alharbi et al. 2013]. Overexpression of HOX genes, such as HOXA5 and HOXC4, can affect differentiation of the myeloid lineage and can lead to proliferation of HSCs [Qu et al. 2014].
CpG island promoter hypermethylation is well recognized in AML [Aggerholm et al. 2006; Deneberg et al. 2010; Paul et al. 2010; Yalcin et al. 2013]. However, methylation of shore, shelf and open-sea regions in AML is less well understood. Qu and colleagues observed increased methylation at CpG island regions in DNMT3A mutated AML, but also hypomethylation patterns at distant shelf and open sea sites [Qu et al. 2014]. DNMT3A mutations have also been shown to be associated with hypomethylation of a small number of genetic loci [Ley et al. 2010]. Considering that a number of studies, as mentioned above, have found that DNMT3A mutations inhibit catalytic activity in a dominant negative manner [Yan et al. 2011; Kim et al. 2013; Russler-Germain et al. 2014], these mutations could in theory be associated with hypomethylation on a global scale.
Ascertaining the cellular consequences of DNMT3A mutations is challenging, however. A number of researchers have noted the lack of correlation between DNA methylation and gene expression in the context of DNMT3A mutations. Changes in DNA methylation outside of promoter regions are likely to play a role [Wu et al. 2010]. There may be other effects via interference with hydroxymethylation or telomere length: DNMT3A null embryonic stem (ES) cells have been shown to have longer telomeres, which may explain the increased replication capacity [Gonzalo et al. 2006; Challen et al. 2012]. Finally, DNMT3A may act within an as yet unidentified protein complex that has altered capacity to function in the absence of DNMT3A [Holz-Schietinger et al. 2012]. Thus, the impact of DNMT3A mutations on DNA methylation is uncertain. It is apparent that such mutations are associated with a distinct methylation signature, demonstrating both hypo and hypermethylation at different sites. Overexpression of HOX genes, reported in a number of studies, is likely to contribute to malignant behaviour in DNMT3A mutated cells. Further investigation into the consequences of this altered DNA methylation signature on gene expression may yield more insights into the leukaemogenic process.
The role of DNMT3A mutations in leukaemogenesis
Perhaps one of the most interesting features of the AML genome is the intriguing clues it may give to the process of leukaemogenesis. DNMT3A mutations are well recognized in myelodysplastic syndrome (MDS) [Walter et al. 2011] and in myeloproliferative neoplasms (MPN) [Viny and Levine, 2014], and persist in AML that develops secondary to these conditions. Their early appearance in such preleukaemic conditions, and persistence throughout leukaemic evolution, suggests that DNMT3A mutations may act as ‘founder’ or initiating genetic lesions. This gives an insight into the role of DNMT3A mutations in de novo AML: it is likely that this mutation occurs very early in the process of leukaemogenesis. This theory is supported by evidence that DNMT3A mutations arise before NPM-1 mutations, which were previously regarded as possible founder mutations. Genetic analysis of 53 AML cases at diagnosis and at relapse revealed that although NPM-1 mutations were initially detectable in a number of cases, they did not always persist when the leukaemia returned [Krönke et al. 2013]. Conversely, DNMT3A mutations present at diagnosis in five cases were also detectable in the relapsed leukaemic cells. This suggests that DNMT3A mutations are present in the majority of subclone populations, and give rise to subclones bearing both mutated and wild-type NPM-1. It is likely that the exact sequence of mutations is not fixed, however, as one case showed loss of the DNMT3A mutation but retained the NPM-1 mutation [Krönke et al. 2013]. DNMT3A mutations may initiate progression towards leukaemia by increasing the likelihood of subsequent mutations, such as FLT3-ITD. AML with DNMT3A mutations is more likely to feature FLT3-ITD mutations at relapse, possibly through a shared pathway linked to the HSC regulator MSI-2 (Musashi-2) [Thol et al. 2013].
Identification of potential founder mutations, such as DNMT3A, may have considerable clinical significance. Genetic analysis of subclone populations in AML have found that leukaemic cells present at relapse are likely to have arisen from a low-level subclone or from an undetected ancestral clone in around 50% of cases, rather than from the dominant clone [Shlush et al. 2014]. Only a small proportion of the mutations identified in AML blasts were identified in HSC from the same patients, and these cells were capable of nonleukaemic differentiation. Shlush and colleagues identify that preleukaemic stem cells have a selective advantage over nonleukaemic stem cells, allowing clonal expansion, but also retain the ability to ‘regenerate the entire haematopoietic hierarchy’ [Shlush et al. 2014]. These cells are found in a high proportion of AML with DNMT3A and IDH2 mutations, and are not depleted by induction chemotherapy, representing a potential reservoir for relapse [Shlush et al. 2014]. These findings highlight a potentially valuable role for the detection and therapeutic targeting of mutations present in early subclones that are also found in relapsed AML, such as DNMT3A. Depletion of the ‘preleukaemic’ stem cell pool may help reduce the risk of later relapse. Moreover, DNMT3A mutations could act as a biomarker in MRD monitoring [Hou et al. 2012].
It is apparent, however, that DNMT3A mutations are not sufficient on their own to cause leukaemia. Mutations in DNMT3A and TET2 have been noted in elderly individuals with nonmalignant proliferation of the myeloid compartment [Busque et al. 2012; Jan et al. 2012], and murine studies have shown that loss of DNMT3A results not in full-blown leukaemia but rather in expansion of the stem cell compartment and some reduction in the ability to differentiate [Challen et al. 2012]. Healthy individuals may have low-level DNMT3A mutations, although it is not clear whether these are acquired mutations or single nucleotide polymorphisms (SNPs), or indeed whether they represent an increased risk of leukaemia. A final, intriguing observation is that of a case report of a donor-derived leukaemia arising in a patient post-HSCT [Yasuda et al. 2014]. The donor bore the same DNMT3A and IDH2 mutations as those detected in the AML cells of the recipient, but remained well. This illustrates the requirement for further mutations to arise to permit full leukaemic transformation, and also suggests that the development of leukaemia is likely to be context-specific.
Prognostic implications of DNMT3A mutations
DNMT3A mutations are consistently associated with a poor overall and relapse-free survival compared with AML with wild-type DNMT3A, although data regarding different subgroups within studies (such as cytogenetic profile and age) are conflicting. Some researchers have found an adverse effect on survival in the non-R882 DNMT3A mutation group, in addition to poorer survival in the younger (<60 years) cohort of R882 mutated AML [Marcucci et al. 2012]. Conversely, other studies have not found an age-dependent variation in prognosis, although this may be a consequence of noninclusion of older age groups in analysis [Ley et al. 2010; Thol et al. 2011]. DNMT3A mutations appear to have varying effects on outcome in different cytogenetic subgroups: Gaidzik and colleagues’ study of 1770 patients suggested a poor prognostic outlook for patients with unfavourable cytogenetics or a mutation in DNMT3A [Gaidzik et al. 2013]. The impact that concurrent molecular mutations may have on prognosis is uncertain, however. Both Thol and colleagues and Ley and colleagues found DNMT3A mutations had an adverse effect in NPM-1 wild type/FLT3 mutated AML [Ley et al. 2010; Thol et al. 2011], while Ribeiro and colleagues found that DNMT3A mutations heralded a particularly poor prognosis when both of the aforementioned genes were wild-type [Ribeiro et al. 2012]. This discrepancy highlights the difficulty inherent to ascribing prognostic influence to a single gene when there are many factors which may influence prognosis, such as the presence of additional ameliorating or exacerbating genes, age, performance status and treatment regimen.
A meta-analysis of 12 cohort studies, including 6377 patients, found that there was a slightly reduced overall survival (OS) across all subgroups, apart from those with favourable risk genetics and non-R882 mutations [Tie et al. 2014]. The meta-analysis noted that there was considerable heterogeneity between reported outcomes in the different studies. This may have arisen from variations on induction chemotherapy regimens, different numbers of patients receiving HSCT and differences in patient characteristics such as age, Eastern Cooperative Oncology Group (ECOG) performance status and white cell count. Another limitation is the inclusion of different types of DNMT3A mutations in the analysis: it is likely that there are biological and prognostic differences between R882 and non-R882 mutations [Marcucci et al. 2012; Gaidzik et al. 2013].
Therapeutic options for DNMT3A mutated AML
DNMT3A mutations have implications for treatment. Patel and colleagues found that patients with DNMT3A, MLL-PTD, and NPM1 mutations have improved survival when treated with intensified dose daunorubicin induction therapy (90 mg/m2 compared with 45 mg/m2) [Patel et al. 2012]. Moreover, other studies have shown an improved survival when patients in this group are treated with intensified dose idarubicin, which, like daunorubicin, belongs to the anthracycline class of chemotherapeutic agents [LaRochelle et al. 2011]. Therefore, there is a likely benefit to the early identification of DNMT3A mutations in patients presenting with AML.
The future of treatment in AML is thought to lie in targeted, personalized therapy. Characterization of pathological processes driving different AML subtypes may identify potential drug targets and consequently improve outcome. Currently, there are no specific targeted therapies aimed at DNMT3A mutated AML. However, epigenetic aberrations are a promising drug target as they are typically reversible. Treatment with demethylating agents, such as decitabine, may provide some therapeutic benefit, which may reflect methylation patterns resulting from altered DNMT3A activity [Metzeler et al. 2012]. High levels of the DNMT3A targeting microRNA, miR-29b, have been associated with improved responses to decitabine in AML patients [Blum et al. 2010]. Research has highlighted micro-RNAs (miRNA) as a potential targeted therapy that could alter DNMT3A gene expression and also act as a potential biomarker. AML blasts have been shown to express a range of miRNAs in different patterns to normal haemopoietic cells [Cammarata et al. 2010]. Micro-RNAs are noncoding RNA molecules that target messenger RNA (mRNA), leading to inhibition of transcription or degradation of the mRNA molecule [Ling et al. 2013]. These miRNA molecules therefore exert considerable control over gene transcription and repression in healthy cells, and may prove useful as a targeted therapy to alter expression of specific genes [Ameres and Zamore, 2013].
Moreover, molecular targets that are not directly related to DNMT3A may have therapeutic value via complex interactions that are not yet fully understood. Bromodomain and extra terminal (BET) family of adaptors contain acetyl-lysine recognition motifs (or bromodomains) that read post-translational acetylation modifications of chromatin (and other proteins modified by acetylation (i.e. nuclear factor kappa B [NFĸB]) [Stewart et al. 2013]. BET bromodomains bind to the acetylated lysine present in the histone tails associated with open chromatin and transcriptional activation. BRD4 is particularly implicated in AML, reading acetylated lysines in histone tails and leading to transcription of key genes concerned with proliferation. JQ1, a potent BRD4 inhibitor, has been shown to inhibit proliferation of the DNMT3A mutated/NPM1c mutated OCI-AML3 cell line [Stewart et al. 2013].
Despite the role of epigenetic abnormalities in malignant haematopoiesis, normal healthy cells also require functional epigenetic machinery. Consequently, treatment directed at epigenetic functions may carry significant risks of off-target drug action. Moreover, as the function of many genes involved in epigenetic regulation is not fully understood, therapies that interfere with epigenetic process may have unpredictable consequences.
Conclusion
DNMT3A is one of the most commonly mutated genes in AML. Although its biology is not yet fully understood, it appears it has a chaotic, Dionysian, effect on methylation patterns, leading to disruption of the orderly, Apollonian, behaviour of healthy bone marrow. As a putative ‘founder’ mutation, DNTM3A mutations set the scene for acquisition of further mutations and the development of AML. The study of DNMT3A provides clues to the process of leukaemogenesis, but also offers valuable practical insights. An example of this is the identification of the relatively anthracycline-resistant behaviour of DNMT3A mutated AML, and the better prognosis afforded by high-intensity induction therapy. This demonstrates the need for a rapid test for DNMT3A mutations: if clinicians were able to detect these mutations soon after presentation, they could ensure that such patients received intensified dose induction chemotherapy. Targeted treatment aimed at ‘founder’ mutations may also deplete the preleukaemic cell reservoir and help reduce the risk of subsequent relapse, thus improving outcomes. Further research into the mechanism underlying DNMT3A mutations has the potential to give rise to more effective, personalized treatments, which may play some role in improving prognosis.
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.