
Abstract
Background:
The phase 3, prospective PROPEL study demonstrated that pharmacokinetic (PK)-guided prophylaxis targeting elevated factor VIII (FVIII) troughs in patients with hemophilia A resulted in lower annualized bleeding rates (ABRs) and a higher proportion of patients experiencing zero bleeds in the second 6 months of treatment when targeting a FVIII trough of 8–12% versus 1–3%.
Objective:
To investigate the benefit of PK-guided prophylaxis with rurioctocog alfa pegol targeting two FVIII trough levels in specific patient subgroups in a post hoc analysis using data from PROPEL.
Design:
This is a post hoc analysis of data from the PROPEL study. The design and primary outcomes of the prospective, randomized PROPEL study (NCT02585960) have been reported previously.
Methods:
This post hoc analysis reports data stratified by FVIII half-life (t1/2), hemophilic arthropathy status, number of target joints at screening, previous treatment regimen, and ABR range in the 12 months before study entry.
Results:
Targeting an elevated FVIII trough of 8–12% was associated with higher average FVIII levels over time, regardless of FVIII t1/2 at baseline. The decrease in total ABR between the 8–12% and 1–3% arms was greatest in patients with a FVIII t1/2 of 6 to <12 h (0.7 versus 3.5); a higher number of target joints, that is, at least four target joints, at baseline (0.2 versus 1.6); the presence of arthropathy (0.1 versus 1.7); and those previously treated on-demand (0.3 versus 1.8).
Conclusion:
These results support the feasibility of targeting elevated FVIII levels using personalized rurioctocog alfa pegol prophylaxis. These benefits may be especially important in patients with a short FVIII t1/2 and those receiving standard prophylaxis with frequent breakthrough bleeds, arthropathy, and target joints.
Registration:
ClinicalTrials.gov Identifier: NCT02585960; https://clinicaltrials.gov/ct2/show/NCT02585960
Introduction
The current standard of care for hemophilia A with a severe phenotype includes intravenous prophylaxis with factor VIII (FVIII) replacement therapy, as well as subcutaneous nonfactor replacement therapy, to prevent bleeding episodes, target joint development, and resultant hemophilic arthropathy. 1 However, patients with hemophilia A still experience high morbidity owing to spontaneous and traumatic bleeding episodes, including into joints and soft tissue.2–4 Standard half-life (t1/2) FVIII treatments require regular infusions at least every 2–3 days to maintain FVIII trough levels for sufficient bleed protection.5–7 Rurioctocog alfa pegol (Adynovate® [US]/Adynov® [Europe]; Takeda Pharmaceuticals U.S.A, Inc., Lexington, MA, USA) is an extended t1/2 recombinant FVIII. It is approved for routine prophylaxis in children and adults in the United States and patients ⩾12 years of age in Europe with hemophilia A.8,9 Clinical data have demonstrated the efficacy and safety of rurioctocog alfa pegol targeting FVIII trough levels ⩾1% in previously treated patients with hemophilia A.10–12 The recommended dose and infusion frequency for routine rurioctocog alfa pegol prophylaxis is a fixed dose of 40–50 IU/kg twice weekly in patients ⩾12 years of age, and 40–60 IU/kg twice weekly in patients <12 years of age.8,9 This reduces the infusion-related treatment burden compared with standard t1/2 products. In addition, extended t1/2 recombinant FVIII products increase the FVIII area under the curve, maintaining FVIII above trough levels for longer periods of time. 13 This may result in improved bleed protection and reduced joint damage.
To further improve the current standard of care, the World Federation of Hemophilia guidelines, along with other groups, recommend using a more personalized and tailored treatment approach. This approach uses prophylaxis to target FVIII trough levels >3–5% on the basis of an individual patient’s pharmacokinetic (PK) profile, bleeding pattern, and lifestyle in patients who do not have satisfactory outcomes with standard prophylaxis.1,14 The phase III, prospective, randomized, open-label, multicenter PROPEL study (NCT02585960) evaluated the safety and efficacy of rurioctocog alfa pegol. 15 This study used PK-guided prophylaxis to target FVIII trough levels of 1–3% or 8–12% to determine the impact of aiming for elevated FVIII levels and a more protective prophylaxis regimen. The primary results of the study according to each treatment arm have recently been reported. 15 The current post hoc analysis of data from the PROPEL study aimed to explore the potential for personalized prophylaxis in specific patient subgroups to identify patients who might benefit the most from a PK-guided treatment approach with rurioctocog alfa pegol to target an elevated FVIII trough. Data from the PROPEL study were stratified by baseline FVIII plasma t1/2, arthropathy status, and joint health as well as treatment regimen and outcomes before study entry.
Methods
Study design and patient population
The PROPEL study, including full eligibility criteria and study design, has been reported previously. 15 The protocol was approved by the independent review board at each participating site, and the study was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice guidelines of the International Council for Harmonization.
In summary, patients were 12–65 years of age with severe hemophilia A (FVIII <1%), with no evidence of FVIII inhibitory antibodies, and an annualized bleeding rate (ABR) of at least two documented and treated bleeds during the 12 months before study entry. Patients had either completed a previous rurioctocog alfa pegol study or were naïve to rurioctocog alfa pegol and had received prophylactic or on-demand treatment with plasma-derived or recombinant FVIII for ⩾150 documented exposure days. After a 72- to 96-h washout period, patients underwent an initial assessment of PK parameters following a single infusion of rurioctocog alfa pegol (60 ± 5 IU/kg). Patients were subsequently randomized to 12 months of PK-guided prophylaxis targeting 1–3% (reference arm) or 8–12% (elevated arm) FVIII trough levels. Randomization was independent of patients’ PK profile. The rurioctocog alfa pegol dose and frequency of administration were based on the patients’ target FVIII trough levels, PK profiles (incremental recovery [IR], plasma t1/2), and actual body mass and could be adjusted for the first 6 months of the study period on the basis of the FVIII trough determined at each study visit.
Study outcome measures
The primary efficacy outcome of the PROPEL study was the presence or absence of any bleeds in the second 6 months of the randomized treatment period (1–3% versus 8–12% FVIII trough levels) and has been previously reported. 15 The current analyses present results from a post hoc evaluation of patient ABRs and the proportion of patients experiencing zero bleeds (total, spontaneous, spontaneous joint, injury related, and injury-related joint) during the second 6 months of the study period in each treatment arm in the per-protocol analysis set (PPAS). These results are stratified by baseline FVIII plasma t1/2, arthropathy status, and presence of target joints, as well as treatment regimen and outcomes before study entry. Baseline and average FVIII levels were measured using the one-stage clotting assay (BCS/XP automated instrument, Siemens, Munich, Germany) with the activator reagent Actin FSL (Siemens, Munich, Germany), and t1/2 was determined from the terminal phase of the concentration-time curve by Phoenix® WinNonlin (Certara, Princeton, NJ, USA). Average FVIII was calculated as time-averaged levels over the course of the study period using predicted FVIII based on dose, IR and t1/2. For the post hoc analyses reported here, data were stratified according to FVIII t1/2 (6 to <12 h, 12 to <18 h, and 18 to <36 h) and hemophilic arthropathy status (yes or no) at screening, treatment regimen and ABR range during FVIII prophylaxis in the 12 months before enrollment (on-demand, prophylaxis with ABR <5, prophylaxis with ABR ⩾5), and number of target joints. As development of the PROPEL study protocol began before the International Society of Thrombosis and Hemostasis definition was published, evaluation of multiple bleeds into the same joint did not follow the currently accepted International Society of Thrombosis and Hemostasis definition of target joint (at least three spontaneous bleeds into a single joint within a consecutive 6-month study period). 16 Therefore, for the purposes of the PROPEL study, target joints were defined as the presence of at least four spontaneous bleeds into a single joint in any consecutive 6-month period in the year before enrollment, and hemophilic arthropathy status was based on the patients’ medical records. Patients’ individual FVIII activity at a given time during the study was predicted using the patients’ dosing information (dose, body mass, date, and time of infusion) and PK parameters (IR and t1/2) before randomization. In addition, prophylactic weight-adjusted consumption of rurioctocog alfa pegol and the frequency of administration were evaluated in each treatment arm in the PPAS according to patient FVIII t1/2 at screening.
Patient ABRs within the 12-month study period stratified according to FVIII activity level during the study are also reported for the full analysis set (FAS).
Statistical analysis
The FAS comprised all patients who were randomized to one of the two prophylactic arms and received at least one dose of rurioctocog alfa pegol. The PPAS comprised all patients in the FAS who completed the full 364-day study period of prophylaxis and had no significant deviations from the protocol that affected the study results. 15 Because of the inherent bias associated with performing a post hoc analysis owing to the potential selection of the analysis based on observed data, the PPAS was chosen for this analysis. The PPAS excludes patients who were not compliant with the protocol, reducing the potential for bias because of noncompliance.
Descriptive statistics were used to analyze the two treatment arms and different patient subgroups. Patient characteristics at baseline were defined for the subgroups used in this post hoc analysis. ABRs and the proportion of patients experiencing zero bleeds were reported using point estimates of the mean and proportion and their 95% confidence intervals (CIs). ABRs were analyzed using separate negative binomial models, including covariates for study arm, ABR range stratum, age, and race. Data analyses for the proportion of patients experiencing zero bleeds during the second 6 months in the two treatment arms were compared using a chi-square test with continuity correction at a two-sided 5% level of significance. All analyses were performed using SAS (SAS Institute Inc, Cary, NC, USA), version 9.4.
Results
Baseline characteristics
The PPAS used for this analysis consisted of the patients who completed the second 6 months of prophylactic treatment and did not have gross protocol deviations (including deviations from informed consent and temperature excursions before investigational product administration) (n = 52, 1–3% reference arm; n = 43, 8–12% elevated arm). 15
Characteristics were similar between the FAS (n = 115) and PPAS (n = 95). All patients were male, with a median (range) age of 29.0 (12–61) years in the PPAS. More than 50% of patients (n = 54) in the PPAS had a FVIII t1/2 between 12 and <18 h at screening. In the PPAS, prophylactic FVIII replacement therapy was previously used by 39 (75.0%) patients in the 1–3% arm and 30 (69.8%) patients in the 8–12% arm. Of the patients who received prior prophylactic treatment, 20 (51.3%) in the 1–3% arm and 16 (53.3%) in the 8–12% arm had an average ABR ⩾5 in the 12 months before enrollment. Overall, 26 (27.4%) patients had no target joints and 69 (72.6%) patients had at least one target joint. The number of patients who had target joints in the 6 months before enrollment was similar across both treatment arms, with the exception of the percentage of patients who had at least four target joints, which was higher in the 8–12% arm. 15 Hemophilic arthropathy was present at screening in 19 (20.0%) patients, seven (36.8%) of whom were in the 1–3% arm and 12 (63.2%) in the 8–12% arm (Table 1). 15
Baseline characteristics (PPAS, N = 95).

FVIII was measured using the one-stage clotting assay and t1/2 was determined from the terminal phase of the concentration-time curve by Phoenix WinNonlin.
Status based on the patient’s medical history (not a determination made by the investigator on the basis of their clinical evaluation of the patient). No other testing was done as part of this protocol to establish arthropathy.
For newly recruited patients, ABRs were assessed on the basis of documented and treated bleeding episodes within 12 months before enrollment.
ABR, annualized bleeding rate; FVIII, factor VIII; PPAS, per-protocol analysis set; t1/2, half-life.
Average FVIII levels
The time-averaged FVIII levels over the second 6-month study period in patients with a target FVIII trough level of either 1–3% or 8–12% stratified by FVIII t1/2 for the FAS are presented in Figure 1. This outcome is influenced by both the patient’s t1/2 and chosen infusion interval. Patients with a target FVIII trough level of 8–12% had a higher average FVIII level compared with those with a target FVIII trough level of 1–3%, regardless of FVIII t1/2. This difference was more pronounced in patients whose baseline FVIII t1/2 was 6 to <12 h.
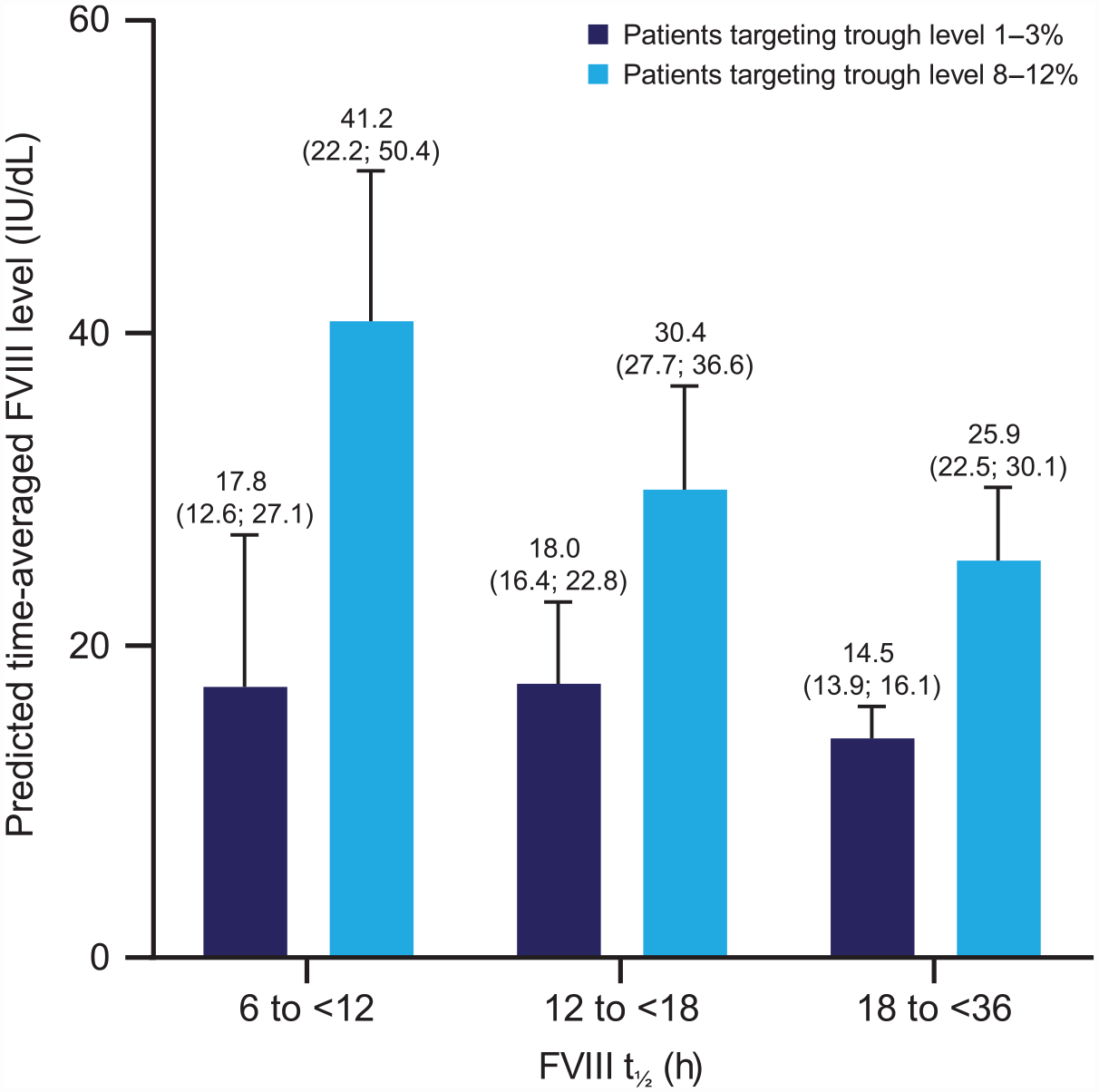
Predicted time-averaged FVIII level stratified by FVIII t1/2 and target FVIII trough level (median [quartile 1; quartile 3]; second 6-month study period; FAS).
Study outcomes
Unless otherwise specified, all of the following results stratified by FVIII baseline t1/2, presence of target joints, presence of arthropathy, previous treatment regimen, and response to treatment refer to the PPAS.
ABRs stratified by baseline plasma FVIII t1/2
Point estimates of the mean (95% CI) for the proportion of patients with zero bleeds and mean ABRs for the 8–12% versus 1–3% arms according to FVIII t1/2 at screening are shown in Table 2. In patients in whom a FVIII trough level of 1–3% was targeted, total ABR decreased, and the proportion of patients experiencing zero total bleeds increased in conjunction with higher FVIII t1/2 at screening. This impact of FVIII t1/2 at screening on bleeding tendency was not present in patients in whom the elevated 8–12% FVIII trough level was targeted. Targeting a FVIII trough of 8–12% also resulted in a higher proportion of patients experiencing zero bleeds and lower bleeding rates for patients with a FVIII t1/2 of 6 to <12 h and 12 to <18 h compared with targeting a trough level of 1–3%. In patients with a FVIII t1/2 of 18 to <36 h, the differences in ABR and proportion of patients with zero bleeds between treatment arms varied by bleeding subtype (total, spontaneous, spontaneous joint, and injury related). Targeting an elevated FVIII trough level of 8–12% was most efficacious in the prevention of spontaneous bleeds, with 71.4–92.3% of patients experiencing zero spontaneous bleeds or spontaneous joint bleeds compared with 41.7–74.2% in the 1–3% arm. There was one patient in the 8–12% arm with a baseline FVIII t1/2 of 18 to <36 h who experienced five injury-related knee bleeds within 6 months.
Proportion of patients experiencing zero bleeds, and mean ABR according to FVIII t1/2 at screening (second 6-month study period; PPAS).

FVIII was measured using the one-stage clotting assay, and t1/2 was determined from the terminal phase of the concentration-time curve by Phoenix WinNonlin.
Point estimates and 95% CIs were obtained from a generalized linear model fitting a negative binomial distribution.
In the 8–12% arm, one patient with a FVIII t1/2 of 18 to <36 h experienced five injury-related knee bleeds in 6 months.
ABR, annualized bleeding rate; CI, confidence interval; FVIII, factor VIII; PPAS, per-protocol analysis set; t1/2, half-life.
ABRs stratified by FVIII activity level
ABRs stratified by FVIII activity level during the 12-month study period (excluding surgery periods; FAS) are shown in Figure 2. Overall FVIII activity levels ⩾20% were associated with lower total, spontaneous, spontaneous joint, and injury-related ABRs compared with FVIII activity levels <20%.

ABR by predicted time-averaged FVIII level (12-month study period; FAS).
ABRs stratified by number of target joints at baseline
Point estimates of mean (95% CI) ABRs for the 1–3% versus 8–12% target arms according to the number of target joints (none, one to three, or at least four joints) are shown in Table 3. Total, spontaneous, and spontaneous joint ABRs were lower in the 8–12% arm compared with the 1–3% arm, irrespective of the number of target joints at screening. Compared with the 1–3% arm, injury-related ABR was slightly higher in the 8–12% arm in patients with no target joints and lower in patients with at least one target joint (Table 3). However, the patient in the 8–12% arm who experienced five injury-related knee bleeds within 6 months was also part of this subgroup.
ABR according to the number of target joints a at screening (second 6-month study period; PPAS).

Target joints were defined as the presence of at least four spontaneous bleeds into a single joint in any consecutive 6-month period before enrollment.
Point estimates and 95% CIs were obtained from a generalized linear model fitting a negative binomial distribution.
In the 8–12% arm, one patient with zero target joints within 6 months before screening experienced five injury-related knee bleeds in 6 months.
ABR, annualized bleeding rate; CI, confidence interval; FVIII, factor VIII; NC, not calculable owing to no events in the 8–12% arm; PPAS, per-protocol analysis set.
ABRs stratified by baseline arthropathy status
Point estimates (95% CI) of the proportion of patients with zero bleeds and mean ABRs for the 8–12% versus the 1–3% treatment arms stratified according to the patients’ arthropathy status at screening are shown in Table 4. A higher proportion of patients had zero bleeds and mean (95% CI) ABRs were lower (total, spontaneous, and spontaneous joint) in the 8–12% compared with the 1–3% treatment arm, irrespective of arthropathy status at screening. In the 8–12% treatment arm, patients with arthropathy at screening had no spontaneous bleeds or spontaneous joint bleeds, and injury-related bleeds were rare (ABR [95% CI], 0.2 [0.02–1.2]). Injury-related ABRs were similar between treatment arms in patients without baseline arthropathy.
Proportion of patients experiencing zero bleeds, and mean ABR according to patient arthropathy status at screening (second 6-month study period; PPAS).
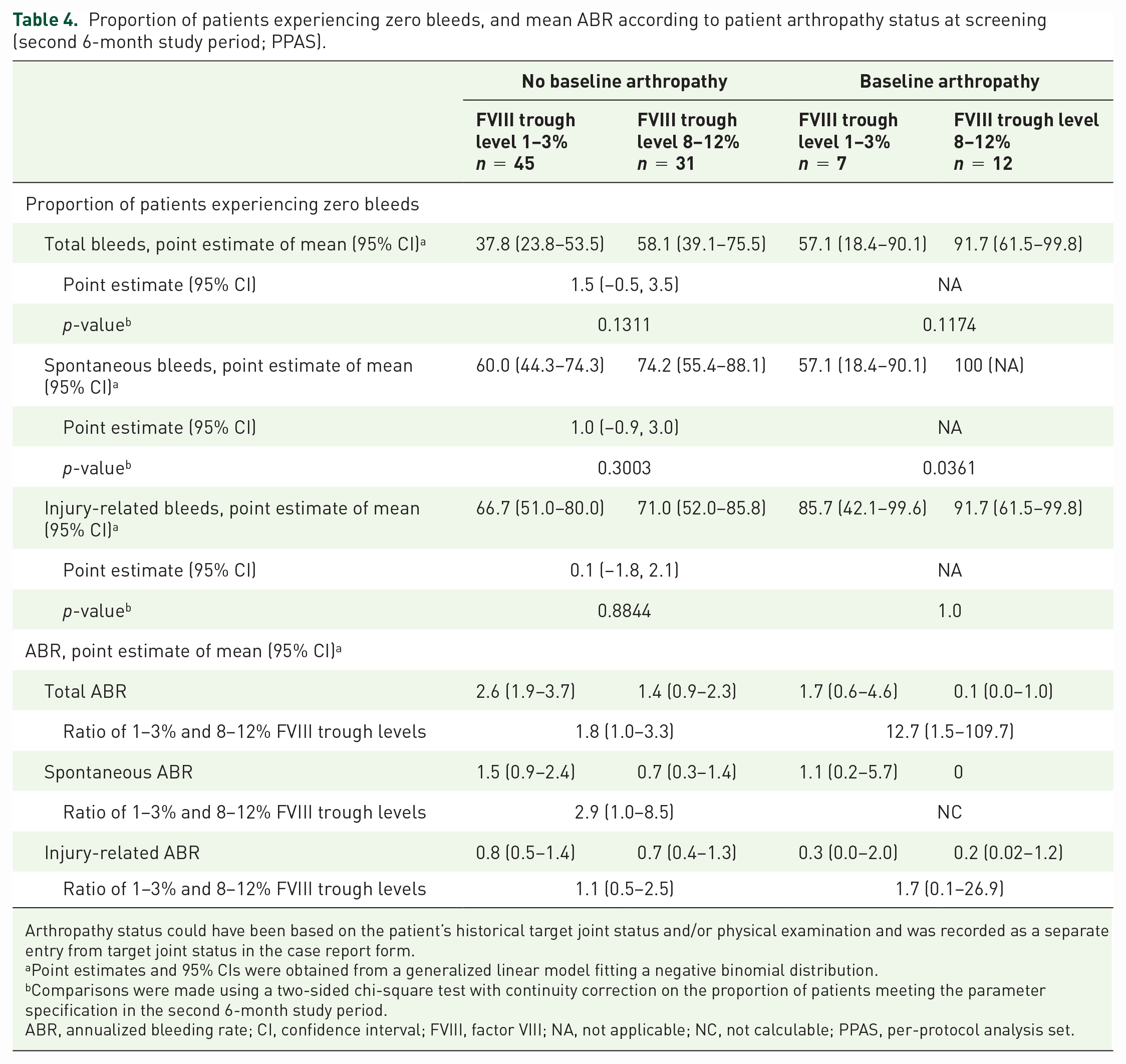
Arthropathy status could have been based on the patient’s historical target joint status and/or physical examination and was recorded as a separate entry from target joint status in the case report form.
Point estimates and 95% CIs were obtained from a generalized linear model fitting a negative binomial distribution.
Comparisons were made using a two-sided chi-square test with continuity correction on the proportion of patients meeting the parameter specification in the second 6-month study period.
ABR, annualized bleeding rate; CI, confidence interval; FVIII, factor VIII; NA, not applicable; NC, not calculable; PPAS, per-protocol analysis set.
ABRs stratified by previous treatment regimen and outcomes
The proportion of patients with zero bleeds and mean ABRs for the 8–12% and 1–3% treatment arms according to the patients’ prior treatment regimens and ABR ranges during FVIII prophylaxis are shown in Table 5. Regardless of prior treatment regimen and ABR range, a higher proportion of patients (⩾50%) in the 8–12% arm had zero total bleeds compared with the 1–3% arm, and their ABRs were lower. Patients previously treated with on-demand FVIII therapy who were assigned to the 8–12% treatment arm during the study had the highest proportion of zero total bleeds (84.6% [95% CI 54.6–98.1]) and experienced no spontaneous joint bleeds. Patients with an average ABR ⩾5 prior to enrollment who were previously treated with prophylaxis also had lower ABRs when treated with rurioctocog alfa pegol targeting a FVIII trough level of 8–12% compared with a trough level of 1–3%. This effect was also observed in patients with an average ABR <5 in prophylaxis prior to enrollment.
Proportion of patients experiencing zero bleeds and mean ABR according to prior treatment regimen and ABR (second 6-month study period; PPAS).

Point estimates and 95% CIs were obtained from a generalized linear model fitting a negative binomial distribution.
Comparisons were made using a two-sided chi-square test with continuity correction on the proportion of patients meeting the parameter specification in the second 6-month study period.
ABR, annualized bleeding rate; CI, confidence interval; FVIII, factor VIII; NA, not applicable; NC, not calculable; PPAS, per-protocol analysis set.
Prophylactic rurioctocog alfa pegol consumption
Weight-adjusted rurioctocog alfa pegol prophylactic weekly dose and dose per infusion according to FVIII t1/2 at screening for the second 6-month study period are shown in Supplemental Table S1. Overall, the weekly prophylactic dose was variable, with overlapping ranges between treatment arms. Mean (standard deviation) prophylactic weekly dose was higher in patients in whom an elevated FVIII trough level was targeted (56.7 [13.9]–201.3 [48.5] compared with 42.3 [11.0]–100.7 [34.9]). The higher baseline FVIII t1/2 was associated with reduced prophylactic dose per week in both treatment arms. A similar trend was observed for the prophylactic dose per infusion, with an even greater overlap between treatment arms and FVIII t1/2 subgroups.
Discussion
The prospective, randomized PROPEL trial was the first study to evaluate the safety and efficacy of PK-guided treatment targeting FVIII trough levels of either 1–3% or 8–12% in patients with severe hemophilia A. 15 This post hoc analysis of the PROPEL study data provides support for treatment using personalized, PK-guided rurioctocog alfa pegol treatment to target elevated FVIII levels in various patient subgroups. This includes patients with a high number of target joints and the presence of arthropathy, as well as those with suboptimal hemostatic efficacy outcomes despite standard prophylaxis.
The efficacy and safety of using a fixed-dose regimen of rurioctocog alfa pegol in patients with severe hemophilia A has previously been demonstrated in various clinical studies.10,11,17 However, fixed-dose regimens do not account for the substantial interpatient variability in FVIII t1/2. Data on the benefits of targeting a FVIII trough level >1–3%, using PK-guided prophylaxis to personalize treatment, are limited and have previously relied on data derived from modeling. 18 In the PROPEL study, targeting an elevated FVIII trough level with the aim of also providing higher and more frequent FVIII peaks with a higher average FVIII concentration was associated with a higher proportion of patients experiencing zero bleeds and consistently lower bleeding rates, with no new safety signals observed. 15 However, the fact that some patients continued to experience bleeding events despite higher target trough levels further underscores the need for truly individualized patient treatment. 19
Data from this post hoc analysis focusing on specific patient subgroups demonstrated that, owing to the patients’ preference for longer infusion intervals, the average FVIII level was higher in patients in whom a FVIII trough level of 8–12% was targeted, regardless of FVIII t1/2 at baseline. This difference was most pronounced in patients with a short (6 to <12 h) FVIII t1/2 (8–12%, 41.2 IU/kg; 1–3%, 17.8 IU/kg). This analysis is also the first to show that targeting a FVIII trough level of 8–12% is associated with improved bleed protection (total, spontaneous, and spontaneous joint) regardless of FVIII t1/2 and arthropathy status at screening. In addition, the impact of targeting an elevated FVIII trough was more pronounced in patients with a short- or mid-range (12 to <18 h) baseline FVIII t1/2, with >90% of patients in whom the higher trough level was targeted experiencing zero spontaneous joint bleeds. The discrepancy between these results and those observed for total ABR and proportion of patients with zero total bleeds in patients with a baseline FVIII t1/2 of 18 to <36 h could be explained by the presence of one patient in the 8–12% arm with a baseline FVIII t1/2 of 18 to <36 h who experienced five injury-related knee bleeds within 6 months. Owing to the relatively low number of patients in this subgroup (n = 7), this patient outlier had a greater impact on the data for both injury-related and total ABR.
These data also demonstrated that higher FVIII activity levels (⩾20%) were associated with lower ABRs, that is, patients rarely bled when their FVIII plasma activity was ⩾20%. The strength of FVIII prophylaxis is that it provides FVIII levels ⩾20%, and these data provide further evidence to support the concept that targeting an elevated FVIII trough leads to higher FVIII levels consistently maintained above this level, which is important to prevent bleeds.
All patients benefited from improved total, spontaneous, and spontaneous joint bleed protection by targeting elevated FVIII troughs, irrespective of the patients’ joint health prior to enrollment. Patients with at least four target joints before screening had the lowest total ABR, and the difference in total ABR between the 8–12% and 1–3% arm was greatest in this group. Injury-related and injury-related joint ABRs were slightly higher in the 8–12% than the 1–3% arm in patients with no target joints before screening, but lower in those with at least one target joint. This discrepancy might be explained by the patient in the 8–12% arm who experienced five injury-related knee bleeds within the 6 months, as this patient had no target joints before screening. Overall, these results suggest that patients with consecutive previous joint bleeds and arthropathy could benefit from targeting of higher FVIII trough levels.
When bleed data were stratified according to prior FVIII treatment regimen (on-demand, prophylaxis with ABR <5 at baseline, and prophylaxis with ABR ⩾5 at baseline), the impact of targeting an elevated trough level was more pronounced in patients with an average ABR <5 treated with prophylaxis prior to enrollment. An ABR ⩾5, despite patients receiving prophylaxis prior to study enrollment, may be indicative of patients who have disease that is more difficult to treat. Although these patients had a reduction in their ABR when treated with rurioctocog alfa pegol targeting FVIII trough levels of 1–3%, reductions were larger when an elevated FVIII trough level of 8–12% was targeted. However, the total ABR (mean [95% CI]) during the second 6 months of the study remained higher than in most of the other treatment subgroups (2.3 [1.3–4.2]). It is also worth noting that, despite having an ABR of ⩾5 at baseline, 50% of patients in whom the elevated trough level was targeted experienced zero total bleeds. Patients previously treated with on-demand therapy in whom the 8–12% FVIII trough was targeted during the study had the highest proportion of zero bleeds (84.6–100.0%). This could potentially be explained if patients receiving on-demand treatment at study entry had a high number of target or problem joints owing to an unsatisfactory treatment model, leading to a disproportionate improvement following the switch to intense prophylactic treatment. In addition, this suggests that initiation of prophylaxis targeting an elevated trough level in patients who previously received on-demand therapy is likely to result in rapid improvement.
Overall, weekly rurioctocog alfa pegol consumption was variable. The overlapping ranges observed between treatment arms likely reflect the heterogeneity of patients’ FVIII t1/2 and emphasize the need for an individualized treatment regimen. In both treatment arms, patients with long FVIII t1/2, and a substantial fraction of patients with mid-range FVIII t1/2, consumed rurioctocog alfa pegol at rates below the recommended weekly dose of 40–50 IU/kg twice per week. All patients in the 8–12% arm with a short FVIII t1/2 consumed more than the recommended weekly dose but had the lowest ABR or best outcome in terms of ABR. The number of infusions required and the potential treatment burden for the patient need to be taken into account when personalizing treatments.
This report has several limitations, including the post hoc nature of the analyses. The number of patients, particularly in certain subgroups, is relatively low, which only allows for descriptive comparisons between the groups. In addition, analysis of safety in the subgroups was not possible due to the lack of power to detect differences, and the use of small subcohorts only allows for descriptive comparisons between the groups. For efficacy analyses, the PPAS was used to reduce the impact of noncompliant patients. The potential for bias must be taken into consideration when interpreting the data. In addition, the PROPEL study population comprised patients with high historical ABR (⩾2), potentially limiting the generalizability of the results to patients receiving prophylaxis with a history of moderate to high ABR. The slightly higher number of patients excluded from the PPAS in the 8–12% arm may also result in limitations in the interpretation of the data obtained from this study. In addition, some bleeds could have been identified on the basis of subjective criteria such as pain consistent with joint bleeds, which was not confirmed by a physician. Therefore, it is possible that patients were not able to differentiate synovitis from the pain associated with a bleed in all cases.
This study and associated post hoc analysis focused on the use of a PK-guided approach to tailor treatment dose and infusion schedule to target specific FVIII trough levels. However, the timing and infusion interval were chosen by the individual investigators. This allows individualization of prophylaxis treatment to ensure peak or high-level FVIII activity during, for example, physical activity. This is very important in clinical practice, where target levels for different patients should also take individual patient lifestyle and physical activity profile into consideration. The ability to optimize treatment in selected groups of patients using different dose regimens without compromising treatment efficacy or patient safety is extremely important in the real-world management of hemophilia A. 20
In conclusion, this post hoc analysis of data from the PROPEL study further supports the feasibility and efficacy of using personalized rurioctocog alfa pegol prophylaxis by targeting elevated FVIII levels with PK-guided dosing. These benefits may be especially important for patients who have experienced multiple joint bleeds and associated joint complications, shorter FVIII t1/2, and those requiring higher FVIII activity levels because of active lifestyles and increased risk of injury-related bleeds. Future studies could aim to confirm this by investigating the use of personalized treatment in these specific patient populations.
Supplemental Material
sj-docx-1-tah-10.1177_20406207231178596 – Supplemental material for Targeting an elevated FVIII level using personalized rurioctocog alfa pegol prophylaxis in specific patient populations with hemophilia A: post hoc subanalysis of the randomized, phase 3 PROPEL study
Supplemental material, sj-docx-1-tah-10.1177_20406207231178596 for Targeting an elevated FVIII level using personalized rurioctocog alfa pegol prophylaxis in specific patient populations with hemophilia A: post hoc subanalysis of the randomized, phase 3 PROPEL study by Carmen Escuriola-Ettingshausen, Robert Klamroth, Miguel Escobar, Oleksandra Stasyshyn, Srilatha Tangada, Werner Engl, Ivan Honauer, Hye-Youn Lee, Pratima Chowdary and Jerzy Windyga in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
The authors thank all patients and their caregivers who took part in the PROPEL study, as well as the study investigators and sites. Under the direction of the authors, medical writing and editorial support was provided by Laura Harrison, PhD, of Excel Medical Affairs (Fairfield, CT, USA) and was funded by Takeda Development Center Americas, Inc., Lexington, MA, USA.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.