
Abstract
Background:
Thrombocytopenia is a common feature of myelofibrosis (MF), a myeloproliferative neoplasm driven by dysregulated JAK/STAT signaling; however, pivotal trials assessing the efficacy of ruxolitinib (a JAK1/2 inhibitor) excluded MF patients with low platelet counts (<100 × 109/L).
Objectives:
Determination of the maximum safe starting dose (MSSD) of ruxolitinib was the primary endpoint, with long-term safety and efficacy as secondary and exploratory endpoints, respectively.
Design:
EXPAND (NCT01317875) was a phase 1b, open-label, ruxolitinib dose-finding study in patients with MF and low platelet counts (50 to <100 × 109/L).
Methods:
Patients were stratified according to baseline platelet count into stratum 1 (S1, 75 to <100 × 109/L) or stratum 2 (S2, 50 to <75 × 109/L). Previous analyses established the MSSD at 10 mg twice daily (bid); long-term results are reported here.
Results:
Of 69 enrolled patients, 38 received ruxolitinib at the MSSD (S1, n = 20; S2, n = 18) and are the focus of this analysis. The incidence of adverse events was consistent with the known safety profile of ruxolitinib, with thrombocytopenia (S1, 50%; S2, 78%) and anemia (S1, 55%; S2, 44%) the most frequently reported adverse events and no new or unexpected safety signals. Substantial clinical benefits were observed for patients in both strata: 50% (10/20) and 67% (12/18) of patients in S1 and S2, respectively, achieved a spleen response (defined as ⩾50% reduction in spleen length from baseline) at any time during the study.
Conclusion:
The final safety and efficacy results from EXPAND support the use of a 10 mg bid starting dose of ruxolitinib in patients with MF and platelet counts 50 to <100 × 109/L.
Registration:
ClinicalTrials.gov NCT01317875.
Introduction
Myelofibrosis (MF) is a clonal, neoplastic disease characterized by bone marrow fibrosis, splenomegaly, and debilitating constitutional symptoms.1–3 The clinical presentation of MF can be highly heterogeneous, but it is typically associated with splenomegaly due to extramedullary hematopoiesis, cytopenias due to progressive bone marrow fibrosis, and constitutional symptoms such as fatigue, night sweats, and fever.4,5 These diverse, disease-associated symptoms negatively impact quality of life and result in reduced life expectancy. MF is driven by dysregulated signaling through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, which is observed in all MF patients, regardless of the presence or absence of JAK2 mutations.6,7
Ruxolitinib is a potent and selective JAK1/JAK2 inhibitor that was approved for the treatment of patients with MF based on its superiority in improving splenomegaly and symptoms, increasing survival and improving quality of life compared with placebo or best available therapy in the phase 3 COMFORT trials.8,9 In these studies, ruxolitinib was generally well tolerated, with adverse events (AEs) rarely leading to treatment discontinuation and managed effectively with dose adjustment.10,11 In these studies, ruxolitinib dosing was titrated according to baseline platelet counts, since efficacy and toxicity are both dose-dependent. 12
As the disease progresses, patients tend to develop thrombocytopenia due to ineffective hematopoiesis and increased splenic sequestration. 13 As many as 26% of patients with MF have low platelet counts (<100 × 109/L). 14 Since the key regulators of thrombopoiesis and erythropoiesis (thrombopoietin and erythropoietin) signal exclusively through JAK2, dose-dependent thrombocytopenia and anemia are expected on-target AEs for patients on ruxolitinib therapy.15,16 Thrombocytopenia is a dose-dependent effect of ruxolitinib and was the dose-limiting toxicity in an initial phase 1/2, dose-finding study of ruxolitinib in patients with MF. 12 For this reason, patients with baseline platelet counts <100 × 109/L have generally been excluded from ruxolitinib clinical trials. In other trials, the ruxolitinib dose was titrated to platelet levels, with 5 mg bid being the current recommended starting dose in patients with platelet counts <100 × 109/L.17,18 Consequently, there are limited data to inform optimal ruxolitinib dosing in patients with MF and lower platelet counts. 19
EXPAND (Evaluating Ruxolitinib in Patients with Low Baseline Platelet Counts Diagnosed With Myelofibrosis) was a single-arm, open-label, dose-finding study of ruxolitinib in patients with primary myelofibrosis (PMF), post-essential thrombocythemia myelofibrosis (PET-MF), and post-polycythemia vera myelofibrosis (PPV-MF), and baseline platelet counts of 50 to <100 × 109/L, which aimed to find the maximum safe starting dose (MSSD) of ruxolitinib and assess its long-term safety and efficacy in this patient population. The MSSD of ruxolitinib was established as 10 mg bid during the 24-week core study period. 20 Through week 48, treatment was generally well tolerated and no new safety signals were detected. Treatment at the MSSD resulted in clinically meaningful spleen size reductions and symptom responses at week 48. 20 After the MSSD had been established, additional patients were enrolled in both strata to assess long-term safety and efficacy during an expansion phase of up to 3 years.
Here, we present the final safety and efficacy results from the 3-year extension phase of the EXPAND study, focusing on patients treated with the MSSD of 10 mg bid. In addition, we also report results from pharmacokinetics/pharmacodynamics (PK/PD) analyses (based on all patients treated in the dose-escalation period) assessing the impact of ruxolitinib plasma levels on platelet count and spleen length response.
Methods
Study design and patient population
EXPAND (NCT01317875) was a phase 1b, open-label, multicenter, dose-finding study of ruxolitinib in patients with MF who had baseline platelet counts between 50 × 109/L and <100 × 109/L, 20 which ran from March 2011 to December 2019. Full details of the study design have been published. 20 Briefly, eligible patients were ⩾18 years of age, diagnosed with intermediate-1, intermediate-2, or high-risk MF (PMF, PPV-MF, or PET-MF), had a palpable spleen (⩾5 cm from the costal margin), and fulfilled platelet count criteria at baseline. Key exclusion criteria included any history of platelet counts <45 × 109/L within 30 days prior to screening, receiving a platelet transfusion within 14 days prior to screening, any history or predisposition to clinically significant bleeding, or any history of platelet dysfunction and/or bleeding diathesis. Based on baseline platelet counts, the patients were assigned to stratum 1 (S1: 75 to <100 × 109/L) or stratum 2 (S2: 50 to <75 × 109/L). The study consisted of a 24-week core study period (including dose escalation and safety expansion phases) to determine the MSSD (incidence rate of dose-limiting toxicity) of ruxolitinib in each stratum and obtain estimates of the efficacy of ruxolitinib in this patient population. This was followed by an extension period beyond week 24 and of up to 3 years, including additionally enrolled patients in both strata, to assess the long-term safety and efficacy of ruxolitinib in patients who received the MSSD (10 mg bid; Supplemental Figure S1). The study ended after all patients had completed their last assessment as per protocol (follow-up visit 30 days after the end of the treatment visit). All data up until the last patient last visit (31 December 2019) are included in the present analysis.
Endpoints
The primary endpoint was the establishment of the MSSD for each stratum during the core study phase, which has been reported previously. 20 This study reports secondary endpoints through year 3, including frequency, duration, and severity of AEs (as per MedDRA version 22.1, CTCAE version 4.03) and serious adverse events (SAEs), spleen response (defined as achievement of ⩾50% reduction in palpable spleen length relative to Day 1 as measured by palpation), assessment of ruxolitinib plasma concentration (using population modeling), and the relationships between the predicted ruxolitinib plasma concentration and PD parameters (platelet counts and palpable spleen length, also using population modeling) over time. Key exploratory objectives included patient-reported outcomes [change in the total symptom score (TSS) as assessed by the modified Myelofibrosis Symptom Assessment Form (MF-SAF) v2.0 diary], and the proportion of patients achieving a ⩾50% reduction in MF-SAF TSS from screening to week 24.
PK assessments and population modeling
To assess the impact of baseline platelet count on the plasma concentration of ruxolitinib, a population PK (PopPK) model previously developed using data from patients with MF and platelet counts ⩾100 × 109/L was validated in patients from the EXPAND study. Ruxolitinib tablets were administered orally twice daily at approximately 12 hours apart. Blood samples for PK analyses were collected for all treated patients (including those treated at doses other than the MSSD, e.g. 5 mg or 15 mg bid) during the dose-escalation period on Days 1, 15, 29, and 57. Samples were collected at a time point between 0.25 and 0.75 hours, 1–3 hours, and 4–12 hours post-dose on Day 1, and pre-dose, 0.25–0.75 hours, and 1–3 hours post-dose on Day 15, with a random time point sample on Days 29 and 57. The plasma concentration of ruxolitinib was determined using a validated liquid chromatography-tandem mass spectrometry assay (LC-MS) with a linear range of 1 to 1000 nM and a limit of quantification of 1 nM.
PD assessment and analysis
The relationship between the estimated ruxolitinib plasma concentration (as predicted by the PopPK model) and platelet counts over time in the EXPAND study was investigated using a PopPKPD model previously developed using data from patients with platelet counts ⩾100 × 109/L. The model was validated with samples taken from EXPAND patients, to confirm the existing relationship was valid within this new population with low platelet counts at baseline. Blood samples for platelet measurements were collected at the times described above for PK assessments, as well as on Day 168 (or the end of treatment visit if earlier than Day 168). Due to the sparse PK sampling collected in this study, average ruxolitinib concentrations (between platelet measurements) were simulated for each patient using the validated PopPK model. In addition, spleen length over time was assessed by quartiles of predicted ruxolitinib plasma concentrations and by ruxolitinib dose level.
Statistics
All patients who received at least one dose of ruxolitinib were included in the analysis. Cumulative data were summarized without separating the core study period and the extension period. The percentage of patients with ⩾50% reduction in spleen length at any time point (including both core and extension periods) and at week 24 were summarized with 95% confidence intervals using Clopper-Pearson exact method.
Ethics
EXPAND was designed and conducted in accordance with the ethical principles of the Declaration of Helsinki, the International Conference on Harmonization (ICH) Harmonized Tripartite Guidelines for Good Clinical Practice, and local laws and regulations. The study protocol and its amendments were reviewed and approved by an independent ethics committee or institutional review board for each study site. Written informed consent was provided by all patients before any study-related procedures commenced. The reporting of this study conforms to the CONSORT statement. 21
Results
Patient disposition
A total of 69 patients were enrolled in the EXPAND study, with 44 patients in S1 and 25 patients in S2. Of these, 38 received ruxolitinib at the MSSD of 10 mg bid (S1, n = 20; S2, n = 18) and are the focus of the safety and efficacy analysis. Overall, 50.0% of patients in S1 and 16.7% of patients in S2 completed the study treatment period as planned. The most common reasons for treatment discontinuation were progressive disease (15%), AEs (10%), and other (10%) in S1, and AEs (33%), death (17%), and physician decision (17%) in S2 (Table 1). The median (range) duration of ruxolitinib exposure was 134.3 weeks (4–210) in S1, and 83.2 weeks (4–161) in S2, with 17/20 patients (85%) in S1 and 11/18 (61%) in S2 receiving ruxolitinib for ⩾48 weeks (Supplemental Tables S1 and S2). Most patients in both strata had at least one dose reduction or interruption, and only 33.3% of patients in the S2 cohort fulfilled drug compliance requirements (relative dose intensity [RDI] ⩾ 83.3%; Supplemental Table S1).
Patient disposition, MSSD cohort.

MSSD, maximum safe starting dose.
Baseline characteristics
Baseline patient characteristics are shown in Table 2. Overall, baseline characteristics were indicative of advanced disease in both strata, although patients in S2 were at a more advanced disease stage than those in S1. Median age was similar across strata, although a larger proportion of patients in S2 were aged 65 or older, compared with S1. The majority of patients in both strata were diagnosed with PMF and had a JAK2 mutation. A large proportion of patients in both strata were classified as intermediate-2 or high-risk MF at screening, but patients with high-risk MF were more common in S2 than in S1. Consistent with a generally more advanced disease stage in S2, median time since initial diagnosis was also considerably longer for patients in S2 compared with those in S1.
Baseline patient characteristics, MSSD cohort.

JAK, Janus kinase; IWG, International Working Group; MF, myelofibrosis; MSSD, maximum safe starting dose; PET, post-essential thrombocythemia; PMF, primary myelofibrosis; PPV, post-polycythemia vera.
Safety
All patients had at least one AE (Supplemental Table S3). SAEs occurred in approximately half of patients across both strata. SAEs considered to be treatment-related occurred in 5/20 (25%) patients in S1 (thrombocytopenia, cardiac arrest, gastrointestinal hemorrhage, drug withdrawal syndrome, meningitis viral, pneumonia, and staphylococcal sepsis) and in 2/18 (11.1%) patients in S2 (thrombocytopenia and pneumonia). The proportion of patients experiencing an AE leading to discontinuation was higher in S2 than in S1, but the proportion of patients reporting AEs leading to dose adjustment or interruption was similar across strata. Cumulative AEs reported in at least 20% of patients are shown in Table 3. The most common AEs in both strata were anemia (S1, all grade, 11/20 [55.0%]; grade ⩾3, 5/20 [25.0%]; S2, all grade, 8/18 [44.4%]; grade ⩾3, 3/18 [16.7%]) and thrombocytopenia (S1, all grade, 10/20 [50.0%]; grade ⩾3, 8/20 [40.0%]; S2, all grade, 14/18 [77.8%]; grade ⩾3, 14/18 [77.8%]). Non-hematological AEs were mostly grade 1 or 2. The most common AE leading to drug discontinuation in both strata was thrombocytopenia (1/20 [5.0%] patients in S1 and 4/18 [22.2%] patients in S2; Supplemental Table S4), which was also the most common AE requiring dose adjustment or treatment interruption (6/20 [30.0%] patients in S1, and 12/18 [66.7%] patients in S2; Supplemental Table S5).
All grade adverse events, regardless of study drug relationship, reported in ⩾20% of patients in either stratum, MSSD cohort.

MSSD, maximum safe starting dose.
A patient with multiple severity grades for an adverse event was only counted under the maximum grade. A patient with multiple occurrences of an adverse event was counted only once in the adverse event category. Adverse events occurring more than 30 days after the discontinuation of study treatment were not summarized.
There were four on-treatment deaths (deaths reported up to 30 days after the last ruxolitinib dose) reported in the MSSD cohort (Table 4). The causes of the two deaths in S1 were cardiac arrest and acute myeloid leukemia, whereas the causes of the two deaths in S2 were multiorgan failure and sepsis. The death due to cardiac arrest occurred 14 days after the last dose of ruxolitinib and was assessed by the investigator as suspected of being related to ruxolitinib; of note, this event occurred in an elderly patient (age 76 years) with an active cardiac condition (left ventricular hypertrophy), concurrent sepsis and acute kidney injury. The other deaths were assessed as being unrelated to study drug.
On-treatment deaths, MSSD cohort.

MSSD, maximum safe starting dose.
Efficacy
A large proportion of patients in both strata achieved clinically meaningful reductions in spleen length (Figure 1 and Supplemental Figure S2A). Among all evaluable patients (i.e. those with both baseline and post-baseline assessment), a spleen response (defined as a ⩾50% reduction in spleen length from baseline) was achieved by 6/15 (40.0%) and 3/8 (37.5%) patients in S1 and S2, respectively, at week 24; 5/15 (33.3%) and 3/10 (30.0%) patients, respectively, at week 48; and by 10/20 (50.0%) and 12/18 (66.7%) patients, respectively, at any time during the study. In terms of symptoms, there was a decrease from baseline – reflecting improvement – in MF-SAF TSS scores, with median (range) changes of −5.3 (−27 to 7) in S1, and −0.1 (–29 to 8) in S2 at week 24 (Supplemental Figure S2B). Treatment with ruxolitinib resulted in improvement in all individual symptoms scored in the MF-SAF TSS form, except for the symptom of bone or muscle pain. At week 24, 30.8% of patients in S1 and 40.0% of patients in S2 achieved a symptom response (⩾50% reduction from baseline in MF-SAF TSS; Supplemental Table S6).
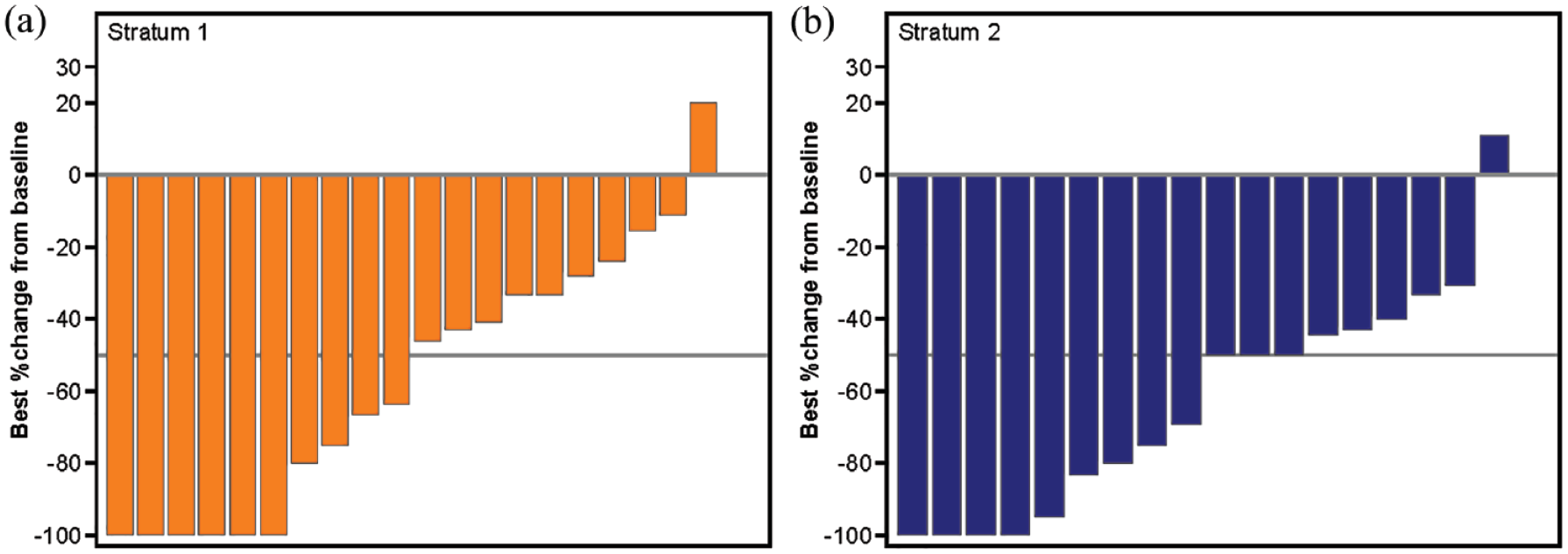
Spleen length response. Waterfall plot of best response in spleen length (with respect to baseline) at any time during the study by stratum, MSSD cohort.
PK/PD population modeling
A total of 276 plasma samples from 40 patients (including patients treated at doses other than the MSSD, e.g. 5 and 15 mg bid) from this study were included to validate a PopPK model previously developed in patients with baseline platelet counts of ⩾100 × 109/L 22 against the EXPAND study population. Overall, modeling results showed that baseline platelet counts had no effect on the PK of ruxolitinib, with no substantial differences in ruxolitinib plasma concentration between patients with baseline platelet counts ⩾100 × 109/L versus those with baseline platelet counts ⩾50 to <100 × 109/L.
An existing PopPKPD model for platelet count versus average ruxolitinib plasma concentration (developed with data from patients with baseline platelet count ⩾100 × 109/L) was validated in the low-platelet population to evaluate its ability to accurately predict platelet count response in patients in the EXPAND study. This was an indirect response model that characterized the effect of ruxolitinib through an inhibitory Emax model applied to the production rate (kin) (the zero-order formation rate of platelets). Platelet counts observed in this study could be fitted well to the model (Supplemental Figure S3), suggesting the relationship between platelet count and predicted ruxolitinib plasma concentration reported in patients with baseline platelet counts ⩾100 × 109/L also applies to this population. However, validation in the low-platelet population showed a slight under prediction of platelet count, particularly at later time points, which may indicate a slightly faster recovery of platelet counts in the low platelet at baseline population compared with patients with normal platelet counts at baseline.
A PopPKPD model also exists to assess the relationship between spleen response and average ruxolitinib plasma concentration; however, this model was based on spleen volume, whereas spleen length (measured by palpation) was assessed in patients in the EXPAND study. Although spleen length is known to correlate well with spleen volume, it was not possible for the previous PopPKPD model to be utilized. Instead, spleen length over time was assessed and stratified by quartiles of predicted average ruxolitinib plasma concentration and by dose level. The results showed that although there was considerable variability in spleen length over the course of the study, an initial reduction in spleen length was clearly observed in the 10 mg bid dose group (Figure 2 and Supplemental Figure S4).

Spleen length over time by ruxolitinib dose. Box plot presentation of spleen length over time by ruxolitinib dosing regimen. Solid black line crosses median values at each time point. Plot shows boxes (25th–75th percentiles) with whiskers (vertical lines) extending to the furthest value within 1.5 × IQR. Dots show outliers.
Discussion
In patients with MF receiving ruxolitinib therapy, current clinical practice is to titrate the dose of ruxolitinib according to the patient’s platelet count; this means that patients with low platelet counts at baseline usually receive a starting dose of 5 mg bid, rather than 15–20 mg bid. However, ruxolitinib response is dose-dependent, 12 suggesting that patients with low platelet counts could potentially be treated with a suboptimal dose.
The EXPAND study aimed to establish the MSSD in both strata (S1, 75 to <100 × 109/L; S2, 50 to <75 × 109/L) and assess the long-term safety and efficacy of ruxolitinib in patients with platelet counts <100 × 109/L. Our results show that a 10 mg bid starting dose of ruxolitinib was well tolerated in a population of patients with MF and low baseline platelet counts (50 to <100 × 109/L). Moreover, ruxolitinib treatment at this dose provided clinically meaningful reductions in spleen length and improvement in clinical symptoms.
The PK/PD assessments further supported the safety and efficacy of ruxolitinib in this patient population: the reduction in platelet count from baseline was similar in patients with low platelet counts compared with those with platelet counts within the normal range at baseline. In addition, a faster platelet recovery was apparent in patients with low platelet counts at baseline, as indicated by under prediction of platelet count at later time points. PK/PD analyses also showed a marked reduction in spleen length for patients treated with a 10 mg bid dose. As such, the MSSD was deemed acceptable in terms of both efficacy and safety.
Evidence from previous clinical trials to inform optimal dosing in patients with platelet counts <100 × 109/L is limited, as these patients were excluded from the pivotal clinical trials COMFORT-I and COMFORT-II which led to the approval of ruxolitinib for patients with MF.10,11 However, a high proportion of patients with MF present with low platelet counts. 14 The more recent JAK inhibitor rUxolitinib in Myelofibrosis Patients (JUMP) study evaluated the safety and efficacy of ruxolitinib in 2,233 patients with MF, including a subset of 138 patients with low platelet counts (<100 × 109/L) at baseline, who were treated with the currently recommended starting dose of 5 mg bid. 18
As expected, a high percentage of patients in EXPAND reported all-grade anemia and thrombocytopenia during the 3-year follow-up (55.0% and 50.0% in S1; 44.4% and 77.8% in S2, respectively), as already reported in the 48-week analysis (45.0% and 40.0% in S1; 44.4% and 77.8% in S2, respectively). 20 These results are in agreement with those from the JUMP study, where the most common AEs in the population with low platelet count at baseline were also anemia (all grade, 52.9% of patients) and thrombocytopenia (all grade, 73.2%). 18 The proportion of patients reporting anemia in the EXPAND study was lower than that reported for patients treated with ruxolitinib in COMFORT-I (96.1%), although more patients in EXPAND reported thrombocytopenia (69.7% in COMFORT-I). 11
In this study, AEs led to treatment discontinuation in 20.0% of patients in S1 and 50.0% of patients in S2, up from 15.0% of patients in S1 and 33.3% of patients in S2 who had discontinued treatment at 48 weeks. 20 Consistent with the 48-week analysis, thrombocytopenia was the most common AE leading to study discontinuation (5.0% in S1 and 22.2% in S2); similarly, in COMFORT-II, thrombocytopenia was the most common AE leading to dose modification (41.0% of patients in the ruxolitinib arm). 10 The low percentage of patients discontinuing treatment due to thrombocytopenia in S1 suggests that this AE is manageable even in this patient population. Although 50.0% of patients in S2 discontinued treatment due to AEs, less than half of these patients did so due to thrombocytopenia. Despite the high incidence of worsening thrombocytopenia in this study, grade ⩾3 hemorrhages were rarely reported in EXPAND patients treated with the 10 mg bid starting dose (three patients, all in S1: one anal hemorrhage, one gastrointestinal hemorrhage and one wound hemorrhage). Furthermore, 20.0% of patients in S1 and 22.2% of patients in S2 remained on treatment for at least 3 years.
As previously reported in the JUMP study, non-hematological AEs were mild, with the most common being grade 1/2 and very rarely leading to treatment discontinuation. Overall, the observed AEs were consistent with the known safety profile of ruxolitinib, and no new or unexpected safety signals were reported. These results support the use of a higher starting dose of ruxolitinib for patients with low platelet counts. As previously mentioned, ruxolitinib toxicity can be managed with dose reductions and interruptions,10,11 and since efficacy is dose-dependent, it is expected that patients will derive a greater benefit from treatment when starting at a higher dose.
One additional on-treatment death (caused by sepsis) was reported among patients receiving the MSSD since the 48-week analysis, leading to a total of four on-treatment deaths reported during the study, with only one case (cardiac arrest, reported during safety follow-up, 14 days after the last dose of ruxolitinib) suspected of being related to treatment.
Ruxolitinib treatment at a starting dose of 10 mg bid provided clinically meaningful reductions in spleen length. A large proportion of patients (50.0% in S1 and 66.7% in S2) achieved a spleen response (⩾50% reduction in spleen length from baseline) at any time during the study; this percentage is higher than that reported at 48 weeks for patients in S1 (40.0%), 20 suggesting that some patients in S1 remaining on treatment for >48 weeks managed to achieve a late spleen response. Response rates were also higher than those reported for patients in the low platelet count subset from the JUMP study (43.8%), 18 who were treated with the 5 mg bid dose. It is important to note that these response rates were observed in the EXPAND study despite dose reductions and interruptions, which affected the majority of patients. The larger proportion of patients in S2 achieving a spleen response could be related to the fact that average spleen length at baseline was larger for this population than for patients in S1.
Results from the JUMP study showed lower rates of spleen and symptom response to ruxolitinib in patients with low platelet counts at baseline compared with the general study population; 18 however, most of these patients remained on the 5 mg starting dose. Analyses of two previous studies17,23 have shown greater clinical improvement following treatment at higher doses. The greater clinical benefits of a higher ruxolitinib dose in patients with MF and low platelet counts are compounded by the manageability and safety of the 10 mg bid dose as shown by our results.
Overall, the safety profile of a 10 mg bid dose of ruxolitinib in this study was acceptable and consistent with the known overall safety profile of ruxolitinib in patients with MF. Treatment at this dose improved symptoms in these patients, even in those at a more advanced stage of disease. Results from this long-term study support the use of 10 mg bid as the maximum recommended starting dose of ruxolitinib in patients with low platelet counts (50 to <100 × 109/L).
Supplemental Material
sj-docx-1-tah-10.1177_20406207221118429 – Supplemental material for Efficacy and safety of ruxolitinib in patients with myelofibrosis and low platelet count (50 × 109/L to <100 × 109/L) at baseline: the final analysis of EXPAND
Supplemental material, sj-docx-1-tah-10.1177_20406207221118429 for Efficacy and safety of ruxolitinib in patients with myelofibrosis and low platelet count (50 × 109/L to <100 × 109/L) at baseline: the final analysis of EXPAND by Paola Guglielmelli, Jean-Jacques Kiladjian, Alessandro M. Vannucchi, Minghui Duan, Haitao Meng, Ling Pan, Guangsheng He, Srdan Verstovsek, Françoise Boyer, Fiorenza Barraco, Dietger Niederwieser, Ester Pungolino, Anna Marina Liberati, Claire Harrison, Pantelia Roussou, Monika Wroclawska, Divyadeep Karumanchi, Karen Sinclair, Peter A.W. te Boekhorst and Heinz Gisslinger in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
The authors would like to thank all patients and their families, all investigators at all study sites, and Vanesa Martinez Lopez, PhD, of Novartis Ireland Ltd, for medical writing support, which was funded by Novartis Pharmaceuticals in accordance with Good Publication Practice (GPP3) guidelines (![]() ).
).
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.