
Abstract
Ruxolitinib became the first US Food and Drug Administration approved therapy for myelofibrosis in 2011 and EU approval is anticipated in summer 2012. Two large phase III trials (known as the COMFORT studies) were the basis for this approval and were published recently. In this review article we discuss the challenges in managing myelofibrosis, the information to date about ruxolitinib and speculate as to the future direction with this and similar agents.
Introduction
The chronic myeloproliferative neoplasms (MPNs) are a group of clonal disorders of a hematopoietic multipotent stem/progenitor cell, initially recognized by William Dameshek in 1951 [Dameshek, 1951]. They include polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). PV and ET can progress to myelofibrosis (MF), referred to as post-PV or post-ET MF, which is clinically indistinguishable from primary MF, and 3% (ET) and 10–15% of cases (PMF) can transform to acute myeloid leukemia (AML) [Vannucchi et al. 2009]. Clinically, they present with variably abnormal blood cell counts (erythrocytosis is predominant in PV and thrombocytosis in ET, while counts may be variably abnormal in MF), splenomegaly, hepatomegaly, foci of extramedullary hematopoiesis, debilitating constitutional symptoms and a propensity to develop thrombotic and hemorrhagic complications.
A major advance in our understanding of the molecular pathogenesis of MPNs occurred in 2005 when the JAK2V617F mutation was described in virtually all patients with PV and around 60% of ET or PMF [Baxter et al. 2005; James et al. 2005; Kralovics et al. 2005; Levine et al. 2005]. Subsequently, several mutations have been described in other genes that to date can be operationally grouped together in three main domains: mutations associated with janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling, mutations affecting genes involved in epigenetic gene regulation and mutations preferentially, though not exclusively, found in association with disease progression or transformation to leukemia. Examples are, respectively, mutations in MPL exon 10, found in around 5–10% of patients with ET or PMF [Pikman et al. 2006; Guglielmelli et al. 2007; Vannucchi et al. 2008; Pardanani et al. 2011c]; JAK2 exon 12 mutations in around 2–3% of V617F-negative patients with PV [Scott et al. 2007; Passamonti et al. 2011], CBL [Grand et al. 2009] and LNK [Oh et al. 2010]; mutations in TET2, EZH2, ASXL1, members of the Polycomb Repressive Complex 2 (PCRC2), SF3B1 and SRSF2 [Carbuccia et al. 2009; Delhommeau et al. 2009; Ernst et al. 2010; Guglielmelli et al. 2011b]. However, mutations in IDH1/IDH2, Ikaros, p53, LNK and DNMT3A have been described preferentially at the time of AML evolution [Green and Beer, 2010; Jager et al. 2010; Pardanani et al. 2010; Abdel-Wahab et al. 2011; Ha and Jeon, 2011; Harutyunyan et al. 2011]. The molecular complexity of MPNs is incompletely understood and represents the focus of an intense research activity.
Of all the MPNs PMF has a more severe course with a median survival of about 6 years, although some patients survive longer than 10 years. Causes of death among 1054 patients with PMF reported by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) were leukemia in 31%, ‘accelerated’ phase without leukemia transformation in 19%, thrombosis in 14%, bleeding in 5%, infections in 10%, portal hypertension in 4%, second neoplasia in 4%, and ‘other’ causes in 10% [Cervantes et al. 2009]. The commonest manifestations of PMF are ascribed to splenomegaly or hepatomegaly, nonhepatosplenic hematopoiesis, thrombohemorrhagic complications and a spectrum of debilitating constitutional symptoms, including severe cachexia, that severely compromise the quality of life in the large majority of patients. Laboratory findings include anemia, thrombocytopenia or thrombocytosis, leukopenia or leukocytosis, and a bone marrow with prominent characteristic megakaryocyte hyperplasia and dysplasia; increased angiogenesis, reticulin and collagen fibrosis as well as osteosclerosis in more advanced disease. These changes in marrow architecture are reactive and largely mediated by an array of locally released cytokines and growth factors, and are slowly but potentially completely reversible after successful stem cell transplantation (SCT). Abnormally increased levels of cytokines, chemokines and proteases are likely also responsible for the systemic manifestation of the disease. The diagnostic criteria for PMF are summarized in Table 1.
Diagnostic criteria for primary myelofibrosis (criteria for post polycythemia vera and post essential thrombocythemia myelofibrosis also exist) [Barosi et al. 2008].
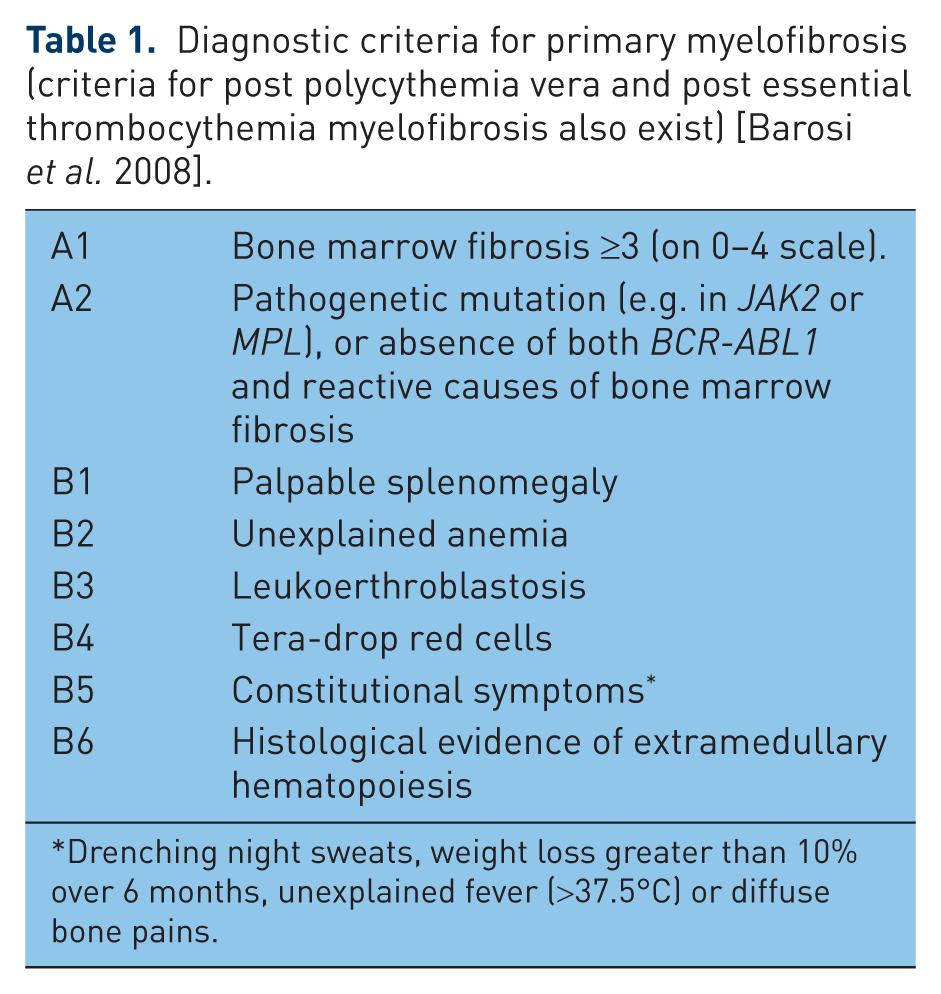
Drenching night sweats, weight loss greater than 10% over 6 months, unexplained fever (>37.5°C) or diffuse bone pains.
With the exception of allogeneic SCT, with its procedure-related mortality, morbidity and failures, no other medical intervention has been shown to have significant impact on the natural course of MF nor to improve survival in patients with MF. Current therapies include cytotoxic drugs (hydroxyurea, busulphan, 2-chlorodeoxyadenosine), erythropoiesis-stimulating agents, androgens, immunomodulators (thalidomide, lenalidomide) and interferon in selected patients with early phase disease; splenectomy is performed in patients with symptomatic splenomegaly or drug-refractory anemia, while radiotherapy is used for sites of extramedullary hematopoiesis. These different options are summarized in Table 2.
Current therapeutic landscape for myelofibrosis.

Risk stratification systems have been devised over the last few years. The International Prognostic Scoring System (IPSS) separates patients with MF into four risk categories (low, intermediate-1, intermediate-2, and high risk) by employing five variables, estimated at diagnosis: age over 65 years, hemoglobin level less than 100 g/l, leukocyte count greater than 25 × 109/l, at least 1% blasts in the peripheral blood, and presence of specific constitutional symptoms (Table 3) [Cervantes et al. 2009]. A dynamic prognostic model (DIPSS) including the same five variables as the IPSS is used to monitor disease progression over time [Passamonti et al. 2010]. Finally, the DIPSS-plus score [Gangat et al. 2011] incorporates three additional variables, ie thrombocytopenia, transfusion dependence and unfavorable karyotype (complex karyotype or sole or two abnormalities such as +8, –7/7q–, i(17q), inv(3), –5/5q–, 12p–, 11q23) for improved prognostic categorization (Table 3).
Current risk score systems for patients with primary myelofibrosis.

Based on the IPSS study, median survival was 135 months [95% confidence interval (CI) 117–181] in the low-risk category (no risk factors), 95 months (95% CI 79–114) in the intermediate-1 (one risk factor), 48 months (95% CI 43–59) in the intermediate-2 (two risk factors) and 27 months (95% CI 23–31) in the high-risk group (three or more risk factors).
DIPSS, dynamic International Prognostic Scoring System; Hb, hemoglobin; IPSS, International Prognostic Scoring System.
The US Food and Drug Administration granted approval of the JAK2 inhibitor ruxolitinib (also known as INCB018424, INC424) as the first in-class drug for the treatment of intermediate and high-risk MF and marked a significant advancement in the therapeutic approach to this incurable disorder. The Committee for Medicinal Products for Human Use gave its approval for this agent which will likely result in full approval for patients with MF and splenomegaly and symptoms. However, the long-term efficacy and the toxicity of ruxolitinib remain to be fully evaluated prospectively.
Identification of a molecular marker for myeloproliferative neoplasms
JAK2 is a nonreceptor tyrosine kinase that transduces signals that originate from several class I membrane cytokine receptors to the cytosol and the nucleus; these mainly include the erythropoietin receptor, the thrombopoietin receptor (that is encoded by MPL) and the granulocyte colony-stimulating factor receptor (G-CSFR). The JAK2 V617F mutation was first described in 2005 [Baxter et al. 2005; James et al. 2005; Kralovics et al. 2005; Levine et al. 2005] and is highly prevalent in MPN.
In vitro and in vivo models indicated that the mutated JAK2V617F allele alone was sufficient to induce the myeloproliferative phenotype in cell lines [Lacout et al. 2006; Wernig et al. 2006; Tiedt et al. 2008] and mice [Bumm et al. 2006; Lacout et al. 2006; Wernig et al. 2006; Zaleskas et al. 2006]. These animals develop an MPN-like phenotype characterized mainly by erythrocytosis or thrombocytosis; the two phenotypes may be largely dependent on the relative ratio of mutant to wild-type JAK2, being lower when thrombocytosis prevails. Marrow fibrosis is usually observed in these mice at later stages, often coincident with decreased erythropoiesis and eventually increased leukocytes, mimicking the transition from a full-born PV to a PPV-MF stage in humans. However, a retroviral MPLW515L model, which also induced JAK activation, produced an acute myeloproliferative disorder with extreme thrombocytosis, bone marrow fibrosis and splenomegaly that rapidly progressed to animal death [Pikman et al. 2006]. These cellular and murine models have been very useful for supporting the rationale of JAK2 inhibition.
Considering the effects of ruxolitinib and other JAK inhibitors on these models of MPN, it was found that the exposure of JAK2V617F overexpressing Ba/F3 cells to the JAK1 and JAK2 inhibitor ruxolitinib resulted in reduced phosphorylation of JAK2, STAT5, and ERK1/2, impairment of cellular proliferation, and induction of apoptosis [Quintas-Cardama et al. 2010]. Furthermore, ruxolitinib suppressed formation of erythroid colonies from MPN primary cell samples with a half maximal inhibitory concentration of 67 nM versus over 400 nM in the case of healthy controls. In a mouse model of JAK2V617F MPN, oral ruxolitinib markedly reduced splenomegaly and circulating levels of inflammatory cytokines, and preferentially eliminated neoplastic cells, resulting in significantly prolonged survival without myelosuppressive or immunosuppressive effects [Quintas-Cardama et al. 2010]. Similar finding were reported for other JAK2 inhibitors, including TG101348 [Wernig et al. 2008], CYT387 [Pardanani et al. 2009b; Tyner et al. 2010], LY2784544 [Florensa et al. 2010; Ma et al. 2010], and leustartinib (CEP701) [Hexner et al. 2008].
Together preclinical models demonstrate reproducible effects of the different JAK2 inhibitors in reduction of splenomegaly and variable control of myeloproliferation, but at the same time established the lack of potentiality of these drugs as concerns the eradication of disease.
Ruxolitinib therapy in myelofibrosis
Phase I and II studies with ruxolitinib (INC424, JAKafi)
A phase I dose-escalation study (study 251) with ruxolitinib commenced very soon after the initial description of the JAK2 V617F mutation [James et al. 2005]. The anticipated dose-limiting toxicity was hematological, relating to nonspecificity of this agent and inhibition of wild-type JAK2 which is essential for normal hemopoiesis. This study was expanded and enrolled a total of 153 patients with median study duration of over 14 months [Verstovsek, 2010a].
In MF the results from this study were unprecedented [Vannucchi, 2010] and clinically can be divided into two main areas for further discussion: splenomegaly responses and symptomatic improvements. Specifically for splenomegaly, the IWG-MRT has defined varying grades of responses in differing aspects of the clinical spectrum that is MF [Tefferi et al. 2006]. According to these criteria, in study 251, 50% of patients achieved an IWG-MRT response graded as ‘clinical improvement’ for splenomegaly; this equates to a 50% reduction in palpable spleen length. In a subset of these patients the investigators evaluated spleen volume by magnetic resonance imaging (MRI), and were able to demonstrate that a median reduction in MRI determined spleen volume of 33% corresponded to median reduction in palpable spleen length of 52%. This facilitated the development of a standard tool for the objective and blinded assessment of response in terms of splenomegaly, and a spleen volume reduction of 35% was later adopted in the phase III studies as the major component of the primary endpoints. Responses for hepatomegaly were more modest. In terms of hematological outcome total leukocyte counts were reduced from 29.8×109/l at baseline to 16.0×109/l after 3 months (p = 0.001), and then subsequently remained stable through 1 year. Sixteen of 17 patients with elevated platelet counts at baseline (mean 728×109/l) had reduced platelet counts at 3 months (mean 336×109/l) [Verstovsek, 2010a].
Whilst reductions in splenomegaly were significant, possibly even more striking results from study 251 were the benefits to patients who had marked and troublesome constitutional symptoms. The performance status for patients on the study was gradually improved and such improvements were generally maintained. This was determined in several ways, for example, 6 min walk tests were performed on 27 patients after one, three, and six cycles of treatment, and the improved mean distances walked were 34, 57, and 71 m. Additionally serial collection of data using a validated MF specific quality of life tool, the MF Symptom Assessment Form (MF-SAF) [Mesa et al. 2009], throughout this trial demonstrated a significant improvement in MF-associated symptoms. The greatest improvements were recorded for abdominal discomfort, night sweats, pruritus and fever. The median weight gain after 1 year in patients receiving ruxolitinib at the identified optimum dose of either 15 or 20 mg twice daily were 9.4 and 7.1 kg, respectively. Furthermore weight gain was more prominent in those with body mass index in the lowest quartile at baseline rather than those in the highest quartile. A reduction in signal transduction and proinflammatory cytokine levels, presumably through JAK1 and JAK2 inhibition, paralleled improvements in the patients’ symptoms (assessed as a reduction in composite symptom score).
Interestingly, but probably not surprisingly, in study 251 results for patients were equivalent irrespective of JAK2 mutational status or subtype of MF (i.e. primary or post-PV MF or post-ET MF). There was only a modest decrease in mutant JAK2 allele burden despite significant clinical benefit, suggesting that the mode of action may be through inhibition of JAK1 signaling and subsequent reduction in inflammatory cytokines, as well as aberrant JAK2 signaling rather than being ascribed to a decrease in allele burden. Indeed data reported for allele burden reduction with ruxolitinib, and other JAK inhibitors must be contrasted with the situation with imatinib and other next generation agents which yield several log reduction in BCR-ABL. Toxicity and safety from the phase I/II and the phase III trials are discussed together below.
Phase III studies with ruxolitinib: the COMFORT trials
Ruxolitinib was subsequently evaluated in phase III trials known as the COMFORT trials (Controlled Myelofibrosis study with Oral JAK Inhibitor Therapy) (Table 4). COMFORT-I was a randomized, double-blind study evaluating the efficacy and safety of ruxolitinib versus placebo in patients with PMF and post-PV/post-ET MF; COMFORT-II, a randomized, open-label study comparing the efficacy, safety, and tolerability of ruxolitinib versus best available therapy (BAT) in patients with PMF and post-PV/post-ET MF. These trials were reported at the American Society of Clinical Oncology and European Hematology Association meetings in 2011, and have recently been published [Harrison et al. 2012; Verstovsek et al. 2012b].
Janus kinase inhibitors currently under development.

Values expressed as Kd (dissociation constant).
Tested only in rheumatologic diseases, not in myeloproliferative neoplasms.
FDA, US Food and Drug Administration; IC50, half maximal inhibitory concentration; JAK, Janus kinase; NA, not available.
COMFORT-I included 309 adult patients with MF randomized 1:1 to ruxolitinib or placebo. Patients in the ruxolitinib arm received 15 mg twice daily (patients with platelet count 100–200 × 109/l) or 20 mg twice daily (patients with platelet count >200 × 109/l). The proportion of patients with spleen volume reduction of at least 35% evaluated by MRI or computed tomography (CT) at week 24 (primary endpoint) was 41.9% with ruxolitinib versus 0.7% with placebo (p < 0.0001) [Verstovsek et al. 2012b]. Indeed the sole responding placebo patient had sustained a splenic infarct, which accounted for the reduced spleen volume, and died shortly thereafter. At week 24, as measured by the modified MF-SAF version 2.0 [Mesa et al. 2011], 45.9% of patients receiving ruxolitinib versus 5.3% of those receiving placebo (p < 0.0001) experienced symptom alleviation with at least 50% reduction in their total symptom score. Mean total symptom score improved by 46.1% in the ruxolitinib arm compared with a worsening of 41.8% in the placebo arm (p < 0.0001). In contrast to the worsening of all individual symptoms observed in the placebo arm, each symptom improved with ruxolitinib treatment (abdominal discomfort, pain under left ribs, early satiety, night sweats, itching, musculoskeletal pain, and inactivity). Quality of life (QOL), measured by European organization for research and treatment of cancer (EORTC) quality-of-life questionnaire core model (QLQ-C30) improved with symptom alleviation. Ten ruxolitinib patients and 14 placebo patients died. The median duration of response and median survival have not yet been reached.
COMFORT-II included 219 adult patients with MF from nine European countries, randomized 2:1 to ruxolitinib or BAT. Patients in the ruxolitinib arm received a starting dose of ruxolitinib of 15 or 20 mg twice daily (as per the COMFORT-I trial), with a possibility of dose titration ranging from 5 to 25 mg twice daily. The proportion of patients with spleen volume reduction at least 35% evaluated by MRI or CT at week 48 (primary endpoint) was 28.5% with ruxolitinib versus 0% with BAT (p < 0.0001) [Harrison et al. 2012]. The proportion of patients with spleen volume reduction of at least 35% evaluated by MRI or CT at week 24 (key secondary endpoint, and equivalent to the primary endpoint of COMFORT I) was 31.9% with ruxolitinib versus 0% with BAT (p < 0.0001). The median duration of response was not reached. Mean improvements from baseline in Functional Assessment of Cancer Therapy–Lymphoma System (FACT-LymS) [Brucker et al. 2005] subscores were greater in the ruxolitinib arm, indicating better QOL versus patients receiving BAT. The EORTC-QLQ-C30 scores for symptoms relevant to patients with MF showed improvement from baseline by week 8 and continued through week 48, also indicating improvement in QOL. Ten and four deaths occurred in the ruxolitinib and BAT arms respectively. The study is ongoing and the results of progression-free survival, leukemia-free survival, overall survival, and change in bone marrow histomorphology are not significantly different between the two arms as yet. Imaging from a patient in COMFORT II is shown in Figure 1 which demonstrates marked durable reduction in splenomegaly and hepatomegaly and also shows weight gain experienced by some of these patients.
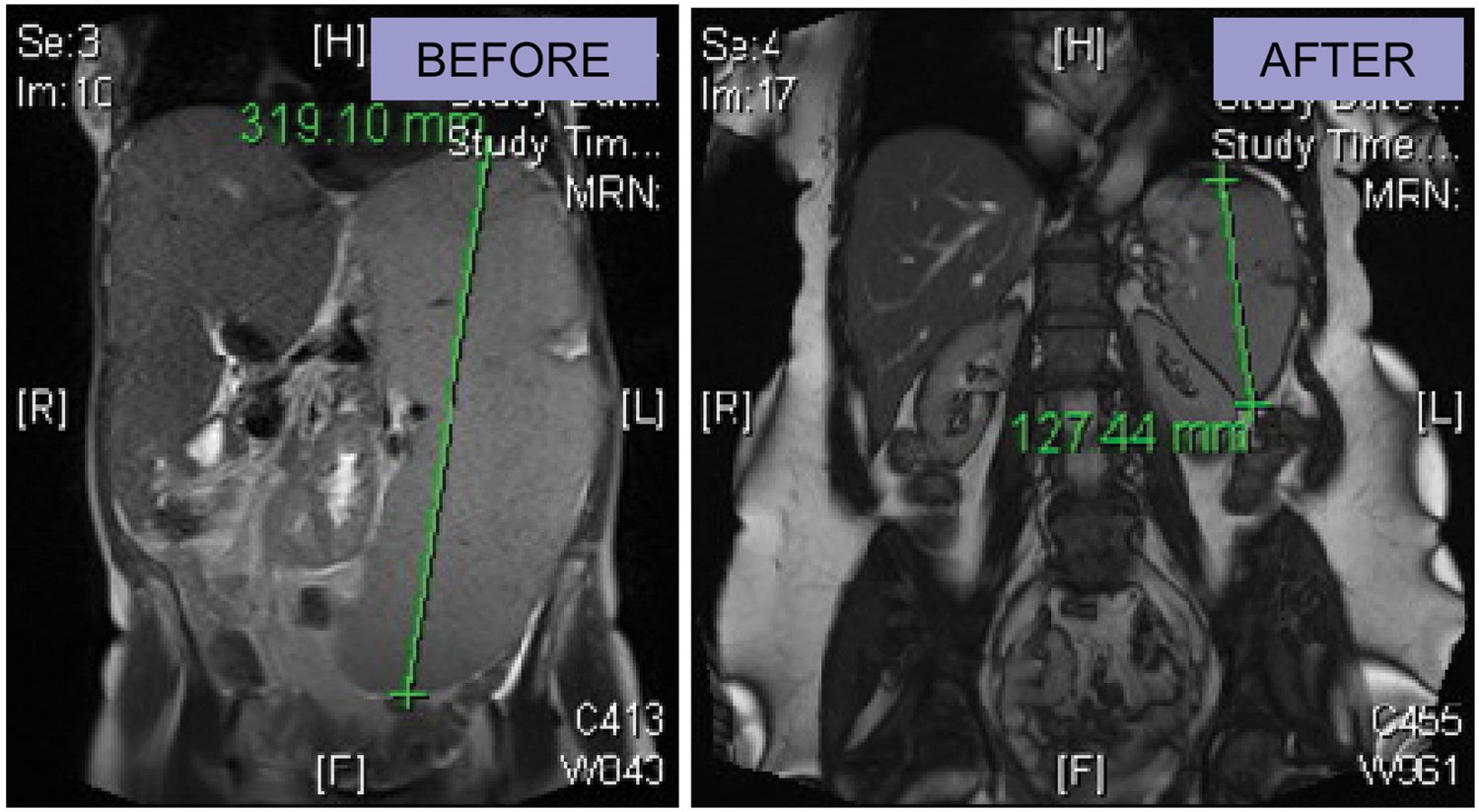
Magnetic resonance imaging from a patient before and after 72 weeks of ruxolitinib therapy.
In the 2011 American Society of Hematology meeting updates from the COMFORT trials [Harrison et al. 2012; Verstovsek et al. 2012b] it was suggested that patients benefitted across all subgroups (PMF, PET-MF, PPV-MF) regardless of JAK2 V617F mutation status, gender or IPSS score [Harrison, 2011; Verstovsek, 2011]. Importantly the COMFORT-I trial after a routine planned safety analysis now shows a clear survival advantage with 13 ruxolitinib and 24 placebo-treated patients dying during the study or during extended follow up (median follow up of 52 and 51 weeks respectively), representing a hazard ratio [95% confidence interval (CI)] of 0.499 (0.254–0.98) (p = 0.0395) for ruxolitinib. For ruxolitinib- and placebo-treated patients respectively, the probability of survival (95% CI) beyond 48 weeks was 0.98 (0.92–0.99) and 0.90 (0.81–0.95) for patients with baseline hemoglobin values at least 10 g/dl and 0.84 (0.72–0.91) and 0.77 (0.63–0.86) for patients with baseline hemoglobin less than 10 g/day [Verstovsek et al. 2011]. This was also shown in a comparison between the MD Anderson cohort of the phase I/II ruxolitinib-treated patients (n = 107) and a matched historical cohort (n = 310) with clinical characteristics that would have allowed them to participate in the phase I/II study of ruxolitinib [Verstovsek et al. 2012a]. The survival of patients with high-risk MF that were treated with ruxolitinib was found to be significantly longer than that of the matched control group (p = 0.005), primarily because of a highly significant advantage in overall survival of patients in the high-risk group (p = 0.006). Intriguingly, there was a significant survival advantage for patients with a greater than 50% reduction in splenomegaly compared with those with less than 25% (p < 0.0001). Further, ruxolitinib therapy was identified as an independent factor influencing better survival in the multivariate analysis. However, an analysis of phase I/II patients treated at the Mayo clinic (n = 51) compared with a collection of patients with MF (all risk groups) did not show similar benefits [Tefferi et al. 2011]. Whether or not there is a true survival benefit with ruxolitinib treatment remains to be assessed in the long term and, preferably, in future ad hoc designed trials; however, overall these data from phase I/II trials suggest the potential of ruxolitinib to ameliorate the natural progression of MF even in patients with advanced disease and would argue strongly for the inclusion of this agent at any early stage into future therapeutic options.
Toxicity and safety of ruxolitinib
In the phase I/II clinical trial thrombocytopenia was reported as the dose-limiting toxicity which was dose dependent and reversible [Verstovsek et al. 2010a]. The likely mechanism of thrombocytopenia is via JAK2 inhibition affecting thrombopoietin signaling, though this has not been fully elucidated. In addition, of patients who were transfusion dependent at baseline, new-onset anemia occurred in 23% and was dose dependent. Hematological toxicity in the placebo-controlled COMFORT-I study revealed grade 3 or 4 anemia in 34.2% and 11.0% of those treated with ruxolitinib and only 15.9% and 3.3% of placebo-treated patients. Interestingly this did not affect response in symptoms or splenomegaly. Significant thrombocytopenia (grade 3 or 4) was seen in 8.0% and 3.9% of ruxolitinib-treated versus 1.3% and 0% of placebo-treated patients. Discontinuation for anemia and thrombocytopenia was rare: only one patient in each treatment group. Neutropenia was less common: 5.2% and 1.9% of ruxolitinib-treated versus 0.7% and 1.3% of placebo-treated patients having grade 3 or 4 neutropenia respectively. These events can be managed by dose reduction or more careful monitoring. Use of ruxolitinib in real clinical practice will require careful monitoring of anemia which usually stabilizes after 12 weeks; dose reduction or hematological support may be required at the clinician’s discretion.
Nonhematologic toxic effects were infrequent and occurred in less than 10% of patients [e.g. asthenia (2.0%), with fatigue, anxiety, fever and insomnia (each 1.3%)]. Two patients (1.3%) with a history of cardiopulmonary disease developed a clinical picture assessed by an investigator as systemic inflammatory response syndrome (SIRS) after abrupt cessation of ruxolitinib. Mayo clinic investigators recently reported five cases developing a SIRS like syndrome on drug withdrawal and a high rate of drug discontinuations, some of these patients were on higher doses of drug than now used in standard practice [Tefferi et al. 2011]. However, a SIRS-like syndrome at the time of reporting has not been observed in the larger phase III COMFORT clinical trials [Harrison et al. 2012; Verstovsek et al. 2012b], which included substantially greater numbers of patients observed for prolonged periods treated at lower doses. The most commonly reported nonhematologic adverse effects of all grades irrespective of causality in COMFORT-I were fatigue (25% versus 34%), diarrhea (23% versus 21%), ecchymosis (19% versus 9%), dizziness (15% versus 7%), and headache (15% versus 5%), and in COMFORT-II diarrhea (23% versus 11%) and peripheral oedema (22% versus 26%). In these trials, discontinuations due to adverse effects were 11% versus 11% (ruxolitinib versus placebo) and 8.2% versus 5.5% (ruxolitinib versus BAT) attesting to the high degree of safety and tolerability of ruxolitinib compared with current agents.
Other JAK2 inhibitors
There are other JAK inhibitors having different on and off target effects, which are at varying stages of development; they are listed in Table 4. It remains to be seen whether any of these agents will be superior to ruxolitinib but some show early promise. For example, CYT387 appears to improve anemia as assessed per IWG response criteria. For the first 60 patients completing at least three cycles of CY387 treatment, responses were reported as 45% spleen, 50% anemia and over 50% constitutional symptoms. A signal was seen for 58% of transfusion-dependent patients becoming transfusion independent for more than 12 weeks. This patient group importantly included patients previously treated with SAR302503, ruxolitinib and pomalidomide [Pardanani et al. 2011a]. SAR302503 may reduce JAK2 V617F allele burden and may in some patients reduce bone marrow fibrosis. After 24 cycles of treatment, the median allele burden was 21% (range 6–100%) compared with 60% (23–100%; p = 0.03) at baseline [Pardanani et al. 2011b]. These data are of significant clinical interest but need to be robustly demonstrated in phase III multicenter randomized trials and the magnitude starkly contrasted with data for BCR-ABL kinase inhibitors such as imatinib and next generation tyrosine kinase inhibitors. For example, such a trial is now open with SAR302503 [ClinicalTrials.gov identifier: NCT01437787].
Current situation and potential for combination therapies
Ruxolitinib has only been tested on a large scale to date in patients with MF with IPSS intermediate risk 2 or above and there is both the rationale and the opportunity to test this agent in patients with earlier stage disease. Such studies are currently underway as are trials in patients with platelet counts below those tested in the phase III trials [ClinicalTrials.gov identifier: NCT01348490]. In addition, since ruxolitinib is so very well tolerated it readily lends itself to being tested in combination. The rationale for combination with other agents could be to improve disease-related responses (for example, with epigenetic therapies) or to use agents which might reduce complications of therapy, for example drug-induced anemia, yet maintaining or even increasing efficacy by allowing higher doses of the drug to be delivered. A summary of such potential agents is shown in Table 5. A further potential benefit of ruxolitinib would be to utilize the agent prior to SCT to improve the patients’ performance status and also significantly to reduce spleen size, since massive splenomegaly (over 22 cm palpable) has been shown to be an adverse factor in predicting transplant outcome [Bacigalupo et al. 2010]. The current role of SCT in MF and the potential additional benefit of ruxolitinib in normalizing inflammatory cytokine profile during and after transplantation is hypothetically attractive and has recently been reviewed [Mclornan et al. 2012].
Potential drug combinations with ruxolitinib.

Information about ‘planned trials’ is to the best of authors’ current knowledge.
BCL, B-cell lymphoma; HSP, heat shock protein; JAK, Janus kinase; MF, myelofibrosis; mTOR, mammalian target of rapamycin; NA, not available; TPO, thrombopoietin.
Unlike for MF, the experience with JAK2 inhibitors in PV and ET is more limited, and few studies have been reported. A phase II trial with ruxolitinib in patients with PV and ET has been completed and preliminary results reported [Verstovsek et al. 2010b]. Here, 39 subjects with ET and 34 with PV who were intolerant/refractory to hydroxyurea according to the European Leukemia Net (ELN) criteria have been included with a median follow up of 10.4 months; additional inclusion criteria were a hematocrit of at least 45% or phlebotomy dependency for PV and platelet count of at least 600 × 109/l for ET. The best dose schedule was determined to be 10 and 25 mg twice daily in patients with PV and ET respectively in portion I of the study. The response to treatment was measured using the standardized ELN criteria [Barosi et al. 2009]. Ruxolitinib led to an overall response rate of 97% (50% complete and 47% partial) in PV and 90% (13% complete and 77% partial) in ET. In PV, 97% of the patients achieved control of hematocrit remaining phlebotomy free, and 68% experienced a complete resolution of enlarged spleen; in more than 70% of patients with leukocytosis or thrombocytosis at baseline, their blood count was normalized. Among patients with ET, 49% achieved a normal platelet count while 79% reached a platelet count less than 600 × 109/l or a decrease greater than 50% at last follow-up visit. In 13 of 14 patients with platelet count greater than 1000 × 109/l, a greater than 50% reduction was observed. Similar to the MF trial, ruxolitinib was well tolerated with a spectrum of toxicity similar to that reported in patients with MF, and included anemia (grade 1–2 in 12% of PV and 18% of ET cases), thrombocytopenia (grade 3–4, 3% of PV cases), and leukopenia (grade 3–4, 5% of PV cases).
The impressive results observed in patients with PV represented the basis for the design of RESPONSE [ClinicalTrials.gov identifier: NCT01243944], a phase III trial.
Use of JAK2 inhibitors in acute leukemia
A phase II study of ruxolitinib in 38 patients with relapsed/refractory leukemia, of whom 18 were secondary leukemias after a previous MPN and 12 had a JAK2V617F mutation, has recently been reported. A dose of 25 mg twice daily, with possible escalation to 50 mg twice daily in case of benefit, was employed, with up to 22, 28-day cycles. Three of the 18 patients showed a significant response, two complete remissions and one partial remission. Overall, the activity of ruxolitinib as a single agent in this patient setting was poor [Eghtedar et al. 2012].
The place of ruxolitinib and other JAK inhibitors in the therapeutic landscape
Ruxolitinib therapy produces remarkable effects on splenomegaly and systemic symptoms, control of leukocytosis and thrombocytosis in most patients, overall improving the quality of life. Whether it truly improved survival is likely to be a matter for debate for some time. The toxicity of this class of drugs, yet with some unique characteristics of the different molecules, appears to be remarkably good, mainly on-target reduction of erythropoiesis and thrombocytopoiesis, which are dose sensitive and rarely produced significant adverse effects, although the transfusion rate increased in a proportion of the patients. Compared with conventional treatments, including splenectomy, JAK2 inhibitors have largely satisfied unmet clinical needs. Thus ruxolitinib undoubtedly represents a highly significant step forward in PMF and other MPN.
This drug is not a parallel with imatinib in chronic myelogenous leukemia. Indeed it is not even necessary to perform a JAK2 V617F test to start therapy with this agent. Subtle differences in activity and toxicity are important to explore in the next generation of JAK inhibitors. A particular area of controversy is the benefit of concurrent JAK1 inhibition that should be assessed in the comparison with more selective JAK2 inhibitors. At the present time treatment with ruxolitinib should be reserved for patients with symptomatic splenomegaly and with severe, life-impairing constitutional symptoms, irrespective of their belonging to risk categories. This would be an effective fit for the likely EU label. Evaluation of other JAK2 inhibitors should be pursued by enrolling suitable patients in novel clinical trials. Another very important opportunity as already discussed is to explore the efficacy of combination therapy, when ruxolitinib is used together with other agents such as histone deacethylase inhibitors, Akt/mammalian target of rapamycin inhibitors, heat shock protein 90 inhibitors, or with erythropoiesis-stimulating agents to compensate for its erythropoiesis-depressing activity, and in the setting of SCT.
Finally, long-term evaluation of toxicity, efficacy, and the impact on overall survival of ruxolitinib and other JAK2 inhibitors is to be established in large prospective series, beyond the information produced by the follow up of patients enrolled in COMFORT-I and COMFORT-II trials. A MF registry (ERNEST) activated as a novel initiative of the ELN to prospectively enroll patients receiving ruxolitinib in European countries, could represent a valuable tool to offer answers to many, still pending, key questions.
Future challenges for therapeutics in primary myelofibrosis
The ERNEST registry offers the potential for us to address several deficiencies in our knowledge about PMF, especially survival and a true estimate of incidence, better understanding about the management of rare events such as Budd Chiari, pregnancy, as well as validating data about prognosis and perhaps refining our current prognostic criteria to include more formally prognosis for PET-MF and PPV-MF. Use of ruxolitinib itself has provoked some further challenges in this field; for example, clearly we should consider revising the IWG-MRT response criteria and look for consensus criteria to define what constitutes progression or resistance to ruxolitinib. Furthermore we need to understand how to better understand the drug’s mode of action to be able to strengthen responses (Box 1).
Key facts about therapy with ruxolitinib
Oral agent with a rapid mode of action
Effective regardless of a patients’ JAK2 V617F status
Significant reduction in spleen volume and troublesome symptoms
Symptoms and splenomegaly recur when therapy is stopped when possible a tapering dose or cover with corticosteroids is recommended
Anemia and thombocytopenia and the on-target effects, these are readily managed by dose reduction
Recent studies suggest a survival advantage may occur with its use, this has yet to be confirmed in later studies and is likely to be due to symptom control
Side effects are rare thus the agent readily lends itself to combination with other therapeutic modalities
Footnotes
Funding
Supported by a grant from Associazione Italiana per la Ricerca sul Cancro (AIRC, Milano) ‘Special Program Molecular Clinical Oncology 5x1000’ to AMV and the AGIMM (AIRC – Gruppo Italiano Malattie Mieloproliferative), project number 1005. A detailed description of the AGIMM project is available at ![]() . This research has also received support from Leukemia and Lymphoma Fund; MPD voice; Novartis; Shire.
. This research has also received support from Leukemia and Lymphoma Fund; MPD voice; Novartis; Shire.
Conflict of interest statement
Alessandro M. Vannucchi have received honoraria for participation to Scientific advisory board by Novartis and Italfarmaco. Claire Harrison is Speaker for Shire, Novartis, Cellgene, Sanofi Avensis.