
Abstract
Background:
In this single-arm phase II study (NCT03557099), we evaluated the efficacy and safety of hetrombopag, a small molecule thrombopoietin (TPO) receptor agonist, in patients with severe aplastic anemia (SAA) who were refractory to standard first-line immunosuppressive therapy (IST).
Methods:
SAA patients who were refractory to standard first-line IST were given hetrombopag orally at an initial dose of 7.5 mg once daily to a maximum of 15 mg once daily, for a total of 52 weeks. The primary endpoint was proportion of patients achieving hematologic responses in ⩾1 lineage at week 18.
Results:
A total of 55 eligible patients were enrolled and received hetrombopag treatment. This study met its primary endpoint, with 23 [41.8%, 95% confidence interval (CI) = 28.7–55.9] patients achieving hematologic response in ⩾1 lineage at week 18 after initiation of hetrombopag treatment. Twenty-four (43.6%, 95% CI = 30.3–57.7) and 27 (49.1%, 95% CI = 35.4–62.9) of the 55 patients responded in ⩾1 lineage at weeks 24 and 52, respectively. Median time to initial hematologic response was 7.9 weeks (range = 2.0–32.1). The responses were durable, with a 12-month relapse-free survival rate of 82.2% (95% CI = 62.2–92.2). Adverse events occurred in 54 (98.2%) patients, and 28 (50.9%) patients had treatment-related adverse events. Seventeen (30.9%) patients had adverse events of grade ⩾3. Serious adverse events occurred in 15 (27.3%) patients and three deaths (5.5%) were reported.
Conclusion:
Hetrombopag showed encouraging efficacy with durable hematologic responses in patients with SAA who were refractory to IST. Hetrombopag was well tolerant and safe for long-term use.
ClinicalTrials.gov identifier:
NCT03557099
Introduction
Severe aplastic anemia (SAA) is a hematologic disorder characterized by bone marrow hypoplasia and peripheral pancytopenia. 1 The majority of SAA cases are thought to be caused by an autoimmune reaction where autologous lymphocytes attack hematopoietic stem and progenitor cells. 2 SAA is often life-threatening due to severe anemia, bleeding, and infection. Immunosuppressive therapy (IST) with antithymocyte globulin and cyclosporine (ATG/CsA) is the standard first-line treatment for patients with SAA who are ineligible for hematopoietic stem cell transplantation (HSCT), and about 70% of patients respond to the treatment.3,4
Management of patients refractory to or relapsed after IST who are ineligible for HSCT is challenging. Limited options of non-transplantation supportive care including transfusion support and anti-infection therapy only help to relieve specific symptoms in the short term. 1 Although retreatment with IST has been effective as salvage therapy in some patients, intensification of the regimen with more potent agents such as rabbit ATG has not improved the response rate as expected. 5 The major long-term concern of SAA is clonal evolution, the risk of which is increased with repeated courses of IST. 6 There was no consensus on the optimal treatment approach for refractory SAA until eltrombopag, the first US FDA-approved (US Food and Drug Administration) oral thrombopoietin (TPO) receptor agonist for patients with SAA who had an insufficient response to IST, with a 40% hematological (platelet, neutrophil, and erythrocyte) response rate at 3–4 months.7,8 Romiplostim, another TPO receptor agonist, which has been authorized for use in patients with refractory SAA in Japan and Korea, led to a hematological response rate at week 27 of 84% [95% confidence interval (CI) = 66%–95%]. 9 However, in several countries including China, neither eltrombopag nor romiplostim has been approved for patients with SAA; therefore, there is an urgent unmet medical need for additional efficient treatment options for patients in these countries.
TPO is a specific regulatory factor that promotes the formation of platelets through binding to its receptor, c-MPL, which is widely expressed in megakaryocytes and hematopoietic stem and progenitor cells.10,11 Previous studies have shown that knockout of c-MPL in a mouse model reduced the number of hematopoietic stem cells, while addition of TPO stimulated the production of hematopoietic stem cells.12–14 This evidence suggests that stimulating c-MPL signaling pathway may initiate hematopoietic-related signaling cascades and induce proliferation and differentiation of bone marrow stem and progenitor cells in patients with SAA.
Hetrombopag is a novel orally bioavailable small molecule thrombopoietin receptor (TPO-R) agonist being developed for the treatment of thrombocytopenia and aplastic anemia. It binds to the transmembrane domain of the TPO-R on progenitor cells and initiates signaling cascades that induce proliferation and differentiation of megakaryocyte progenitor cells, leading to increased platelet production. 15 Hetrombopag has a similar mechanism of action in stimulating the TPO-R signaling pathway as eltrombopag and showed better pharmacological performance both in vitro and in vivo (nude mice) in the preclinical study. 16 In phase I trials, hetrombopag was safe and well tolerated in healthy participants with acceptable pharmacokinetic profiles.17,18 In a phase III trial conducted in patients with immune thrombocytopenia (ITP) who had not responded to or had relapsed after previous treatment, hetrombopag was shown to have effective thrombopoietic activity, and the hematological response rates were significantly higher both in the hetrombopag 2.5 mg group [58.9%; odds ratio (OR) = 25.97, p < 0.0001] and 5 mg group (64.3%; OR = 32.81, p < 0.0001) than in the placebo group (5.9%) at week 8; 19 a long-term treatment with hetrombopag for up to 48 weeks was effective in increasing and maintaining platelet count within the desired range in adults with ITP, and hetrombopag was well tolerated. 20 This trial provided clinical evidence of the platelet-enhancing potency of hetrombopag.
In this context, we conducted a phase II trial to assess the efficacy and safety of hetrombopag in patients with SAA who were refractory to IST. Based on the data from this study, hetrombopag was approved by the China National Medical Products Administration on 11 June 2021, for use in patients with SAA who are refractory to IST. Here, we report the results of this trial.
Methods
Study design and participants
In this open-label, single-arm, phase II study which included core period and extension period, we enrolled patients from 14 clinical centers or hospitals in China (Supplemental Table S1). Eligible patients were 18 years or older, had a diagnosis of SAA according to the Camitta criteria and thrombocytopenia (defined as platelet count ⩽30 × 109/l),21,22 had an insufficient response to at least one course (6 months) of previous ATG-based IST, and ineligible for or unwilling to undergo transplantation. Exclusion criteria included previous IST treatment within 6 months of enrollment, a history of treatment with TPO-R agonists such as eltrombopag or romiplostim (except TPIAO, a recombinant human TPO) within 3 months, bleeding or infection not responding to appropriate therapy, paroxysmal nocturnal hemoglobinuria (PNH) clone size in neutrophils of ⩾50%, human immunodeficiency virus (HIV), hepatitis B virus (HBV) or hepatitis C virus (HCV) positivity, Eastern Cooperative Oncology Group (ECOG) performance status of 3 or greater, the occurrence of hematologic malignancies or solid tumor within 5 years, cytogenetic abnormalities, congestive heart failure, arrhythmias, or arterial/venous thrombosis requiring treatment within 1 year or myocardial infarction within 3 months.
The trial was conducted in accordance with Declaration of Helsinki and Good Clinical Practice Guidelines. The protocol and all amendments were approved by the institutional review board or independent ethics committee of each participating clinical center (Supplemental Table S1). All patients provided written informed consent. This trial is registered with ClinicalTrials.gov, identifier NCT03557099. The reporting of this study conforms to STROBE (Strengthening the Reporting of Observational studies in Epidemiology) guidelines. 23
Procedures
During the 18-week core period, 55 eligible patients were given hetrombopag orally at an initial dose of 7.5 mg once daily after overnight fasting.17,18 If, after 2 weeks, the platelet count had not increased by 20 × 109/l from baseline and toxicity was tolerated, the dose of hetrombopag was gradually up-titrated in single steps by 2.5 mg every 2 weeks to a maximum of 15 mg once daily. Patients were followed up every 2 weeks. Dose reduction or treatment interruption/discontinuation was permitted according to efficacy and safety. If a patient had an increase in platelet count of ⩾20 × 109/l but the total platelet count was ⩽200 × 109/l, the current dose was not changed. If a patient had a platelet count of 200 × 109/l–400 × 109/l, hetrombopag was reduced by 2.5 mg. If the platelet count reached >400 × 109/l, hetrombopag treatment was suspended and an evaluation was done 1 week later. Once the platelet count decreased to ⩽200 × 109/l, the treatment was resumed at a dose 2.5 mg lower than previously administrated. If the platelet count remained at a level >400 × 109/l after 2 weeks of treatment at the lowest dose, hetrombopag was discontinued. A Bayesian stopping rule for safety was applied after 20 and 40 patients were treated with hetrombopag to monitor treatment-related serious adverse events, including death, any grade 4 toxicity, and grade 4 thrombosis/embolism. 24
The extension period was divided into two stages. After completion of treatment in the core period, all patients voluntarily could choose to enter the first stage (weeks 19–24) of the extension period. Patients who completed the first stage and achieved a hematological response to hetrombopag could voluntarily enter the second stage (weeks 25–52), while patients with no hematological response to hetrombopag were withdrawn from the study. Patients were followed up every 2 weeks in the first stage and every 4 weeks in the second stage.
Assessments
Platelet, erythroid, and neutrophil response were evaluated at weeks 18, 24, and 52 after initiation of hetrombopag treatment. Platelet response was defined as an increase of ⩾20 × 109/l from baseline or independence from platelet transfusions for ⩾8 weeks in patients who were previously transfusion-dependent. Erythroid response was defined as an increase in hemoglobin by ⩾1.5 g/dl from baseline, a reduction of ⩾4 transfusion units for 8 weeks compared with transfusion requirements during the 8 weeks before enrollment, or independence from red cell transfusions for ⩾8 weeks in patients who were previously transfusion-dependent. Neutrophil response was defined as an increase in neutrophil count by >100% or by >0.5 × 109/l from baseline or an independence from granulocyte colony-stimulating factor (G-CSF) for ⩾2 weeks in patients who were previously G-CSF-dependent. However, improvements in platelets, hemoglobin, or neutrophils that relied on exogenous transfusions or injections were not considered as meeting the response criteria. Hematologic response was defined as fulfilling at least one lineage of platelet, erythroid, or neutrophil response. Relapse was defined as meeting the diagnostic criteria for SAA again (at least two of the following criteria: neutrophil count, <0.5 × 109/l, platelet count, <20 × 109/l, and absolute reticulocyte count, <20 × 109/l). Chromosome karyotype analyses were performed at baseline, 18 weeks, and 52 weeks after hetrombopag administration to detect clonal evolution. Bone marrow examinations were performed at baseline, week 18, and before leaving the study.
Adverse events were recorded starting at the signing of the informed consent and ending 28 days after administration of the last dose, and graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
Outcomes
The primary endpoint was proportion of patients achieving hematologic response at week 18. Secondary endpoints were proportion of patients achieving hematologic response at week 24 or 52; proportion of patients achieving bilineage response, trilineage response, platelet response, erythroid response, or neutrophil response at week 18, 24, or 52; proportion of patients becoming independence from platelet transfusion, red blood cell transfusion, or G-CSF injection; and reduction of ⩾4 transfusion units of red blood cells for 8 weeks compared with transfusion requirements before enrollment.
Statistical analysis
Sample size was calculated based on the primary endpoint (the proportion of patients with hematologic response in at least one lineage at week 18). The exact probability method assumes a response rate of 10% as the null hypothesis and a one-sided significance level of 0.025. In order to detect a response rate of 30% in patients treated with hetrombopag, a sample size of 41 patients provides a power of 90%. Considering a drop-out rate of 10% or higher, this study planned to enroll 55 patients.
Full analysis set (FAS), the primary population for efficacy analysis, included all enrolled patients who received at least one dose of hetrombopag and had at least one assessment after baseline. The safety set included all patients who received at least one dose of hetrombopag treatment. The primary endpoint was reported with 95% CIs, calculated with Clopper–Pearson binomial CIs. Time-to-event data were estimated using Kaplan–Meier analysis and presented with accompanying 95% CI. Patients without hematologic assessment at week 18 were calculated as non-responder. For continuous variables, means were compared using two-sample t test; for the comparison of categorical variables, chi-square test was used. All analyses were conducted with SAS software, version 9.4.
Results
Patients
Between 20 June 2018 and 24 May 2019, 74 patients were screened and 55 eligible patients received hetrombopag treatment (Figure 1). Patients’ baseline characteristics and treatment history are presented in Table 1. As of data cutoff on 12 June 2020, the median duration of follow-up was 12.9 months [interquartile range (IQR) = 5.7–13.8]. No patient remained in the study. Of the 55 patients enrolled in the study, 49 (89.1%) completed treatment in the 18-week core period. A total of 41 (74.5%) patients completed 24 weeks of treatment in the extension period stage 1, and 30 (54.5%) completed 52 weeks of treatment in the extension period stage 2. Twenty-five patients (45.5%) did not complete the 52-week study period, the primary reasons being patient decision (8/55, 14.5%) and lack of efficacy (8/55, 14.5%).
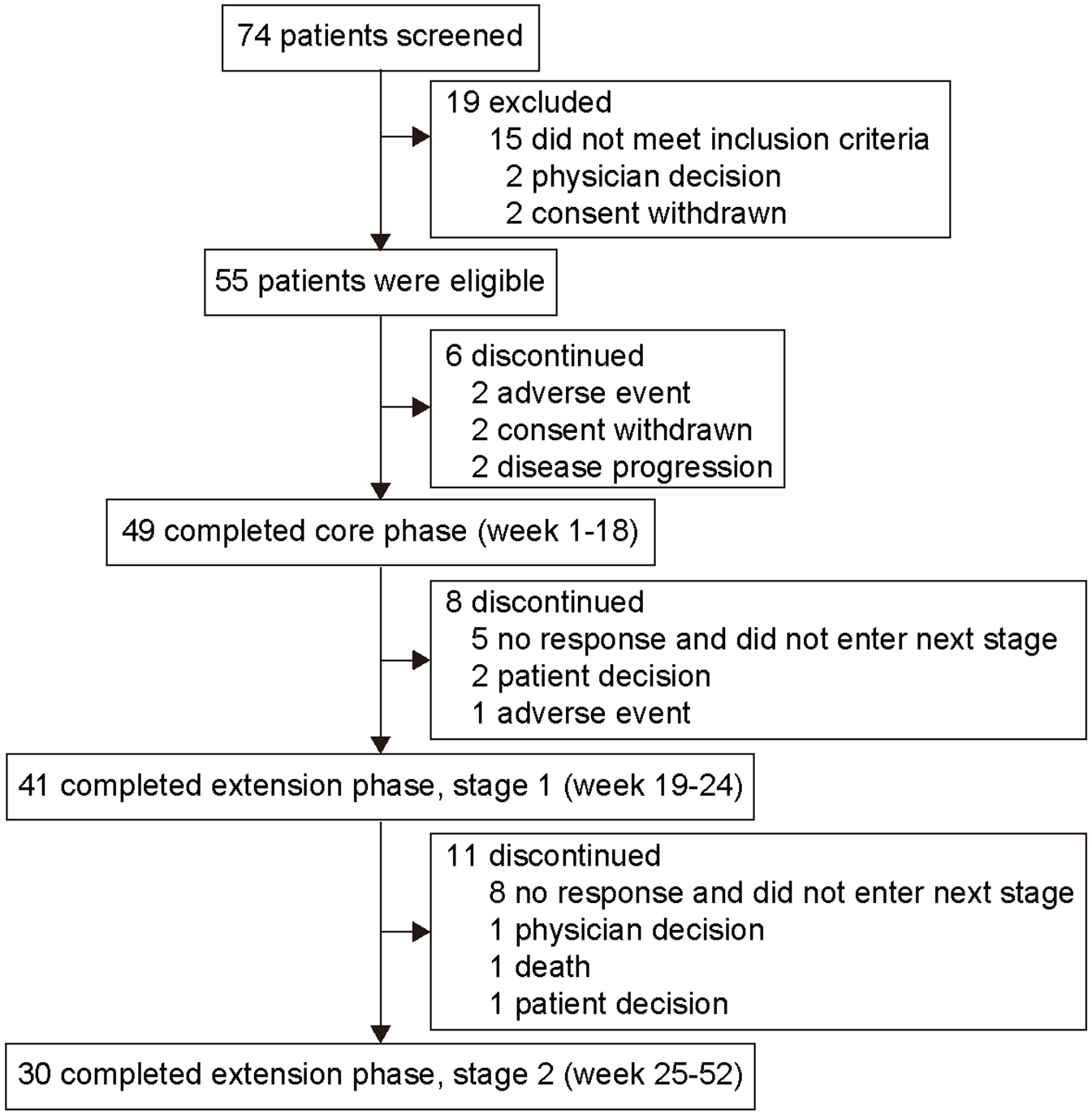
Study profile.
Patient characteristics and treatment history.
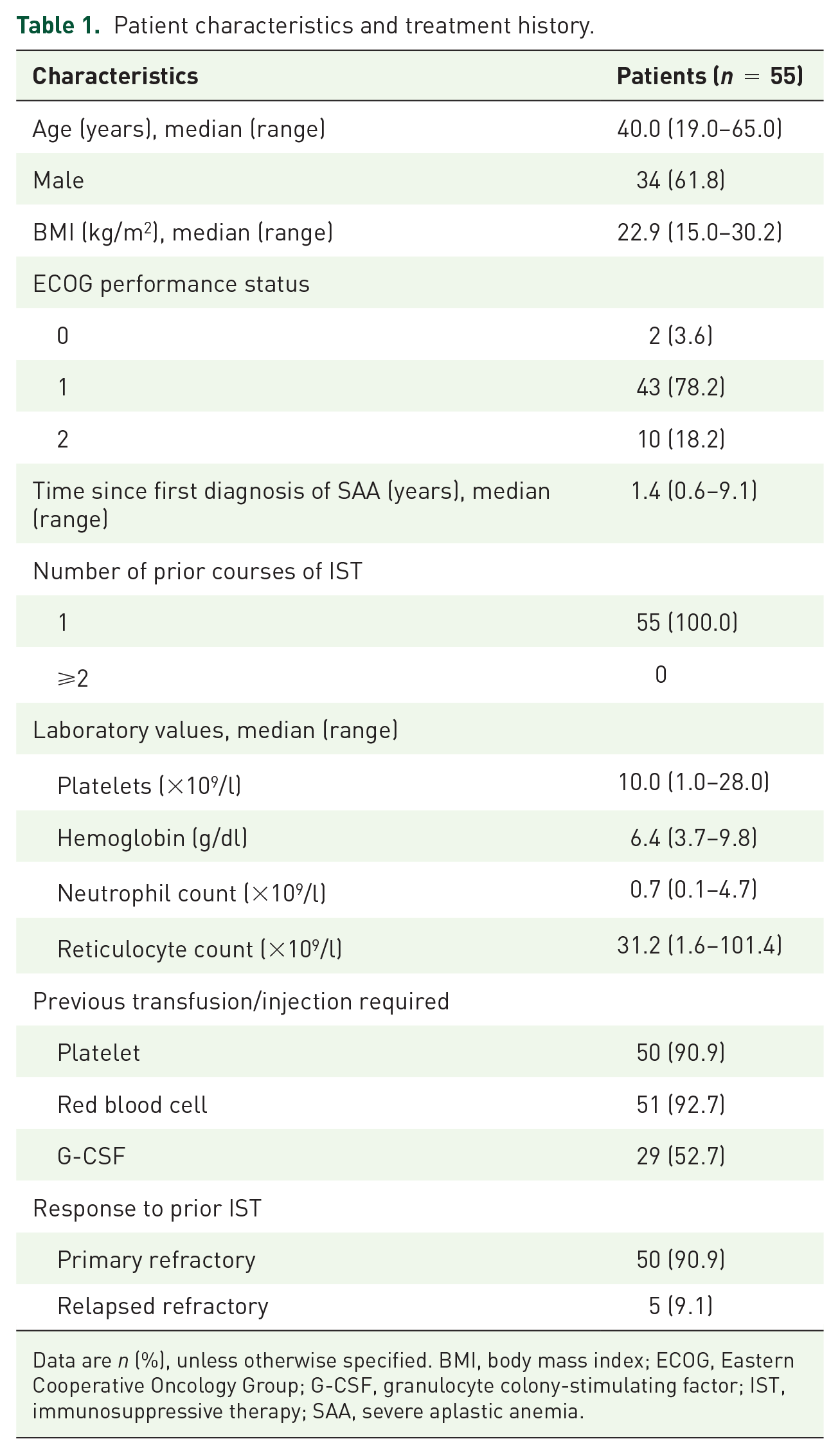
Data are n (%), unless otherwise specified. BMI, body mass index; ECOG, Eastern Cooperative Oncology Group; G-CSF, granulocyte colony-stimulating factor; IST, immunosuppressive therapy; SAA, severe aplastic anemia.
As of data cutoff, 52 (94.5%) patients were titrated to the maximum dose of 15 mg once daily. The final dose of the three patients who did not complete the study was 7.5, 10, and 12.5 mg. The median exposure period to hetrombopag was 51.6 weeks (range = 0.1–53.1).
Efficacy
This study met its primary endpoint, with 23 (41.8%, 95% CI = 28.7–55.9) of the 55 enrolled patients achieving hematologic response at week 18 after initiation of treatment (Figure 2 (a)). Hematologic responses in subgroups were generally consistent with the primary finding, with the lower limits of their 95% CIs greater than 10% (Supplemental Figure S1). The proportion of patients who achieved platelet response, erythroid response, and neutrophil response at week 18 was 14.5%, 34.5%, and 25.5%, respectively (Table 2). Regarding predictors of response (Supplemental Table S2), hematological response was significantly positively correlated with platelet and reticulocyte count, but not correlated with age, neutrophil count, time since first diagnosis of SAA, or PNH clone.

Lineage characteristics of hematologic responses to hetrombopag. The Venn diagrams show the number of patients with unilineage, bilineage, and trilineage hematologic responses at week (a) week 18, (b) week 24 and (c) 52
Proportions of patients requiring platelet, red blood cell, or G-CSF transfusion/injection over time after hetrombopag treatment.

Data are n (%, 95% CI). ‘Independence’ was defined as for patients who were dependent on platelet/red blood cell transfusion or G-CSF injection before enrollment, they should have stopped platelet transfusion for at least 8 or 2 consecutive weeks at week 18, 24, or 52. CI, confidence interval; G-CSF, granulocyte colony-stimulating factor.
A total of 44 (80.0%) of the 55 patients enrolled in the study entered the extension period stage 1 and continued to receive hetrombopag during weeks 19–24 of treatment. Twenty of these patients maintained hematological responses from week 18 to week 24. Together with four patients who obtained the first hematologic response at week 24, the response rate of the total population at week 24 was 43.6% (24/55, 95% CI = 30.3–57.7, Figure 2(b)). Thirty-three (33/55, 60.0%) patients entered the extension period stage 2 (weeks 25–52) to continue hetrombopag treatment, and at the 52-week evaluation, a hematologic response occurred in 27 patients (27/55, 49.1%, 95% CI = 35.4–62.9; Figure 2(c)).
Among the 31 patients achieving hematologic response at week 18, 24, or 52 from the first day of dosing, the median time to the first hematologic response was 7.9 weeks (range = 2.0–32.1). The response was maintained in 26 (83.9%) patients, while 5 (16.1%) developed disease relapse. The swimmer plot showed that three out of the five patients who relapsed achieved a hematologic response again after continuing hetrombopag treatment (Supplemental Figure S2). The median duration of hematologic response has not yet been reached due to the small number of patients with relapse. The Kaplan–Meier estimated probability of relapse-free survival at 12 months was 82.2% (95% CI = 62.2–92.2; Supplemental Figure S3).
The proportion of patients who achieved platelet response, erythroid response, or neutrophil response also increased gradually with the continuation of treatment (Figure 2(a)), and at week 52, the proportions were 27.3%, 40.0%, and 40.0%, respectively (Table 2). The proportion of patients achieving trilineage response was 10.9% (6/55, 95% CI = 4.1–22.2) at week 18, which increased to 16.4% (9/55, 95% CI = 7.8–28.8) at week 24 and 21.8% (12/55, 95% CI = 11.8–35.0) at week 52.
The platelet, hemoglobin, neutrophil, and reticulocyte counts all progressively increased over the 52-week period, and their mean change from baseline over time is shown in Figure 3. For platelet counts, the mean [standard deviation (SD)] change from baseline was 10.0 (19.1) × 109/l, 14.1 (27.9) × 109/l, and 38.2 (54.6) × 109/l at weeks 18, 24, and 52, respectively. The mean (SD) change in hemoglobin from baseline was 1.1 (2.3) g/dl (week 18), 1.8 (2.4) g/dl (week 24), and 3.5 (2.7) g/dl (week 52). Similarly, the mean change in neutrophil count increased over time, with 0.3 (1.1) × 109/l (18 weeks), 0.3 (1.2) × 109/l (24 weeks), and 0.8 (1.1) × 109/l (52 weeks) as did the reticulocyte count, 7.3 (21.2) × 109/l, 12.4 (24.0) × 109/l, and 24.1 (25.6) × 109/l at weeks 18, 24, and 52, respectively. In addition, the proportion of patients requiring platelet transfusion, red blood cell transfusion, or G-CSF injection all gradually decreased over time after hetrombopag treatment. As shown in Table 2, the proportions of patients at 52 weeks who were platelet, red blood cell, and G-CSF transfusion/injection-independent were 18.0%, 27.5%, and 34.5%, respectively.
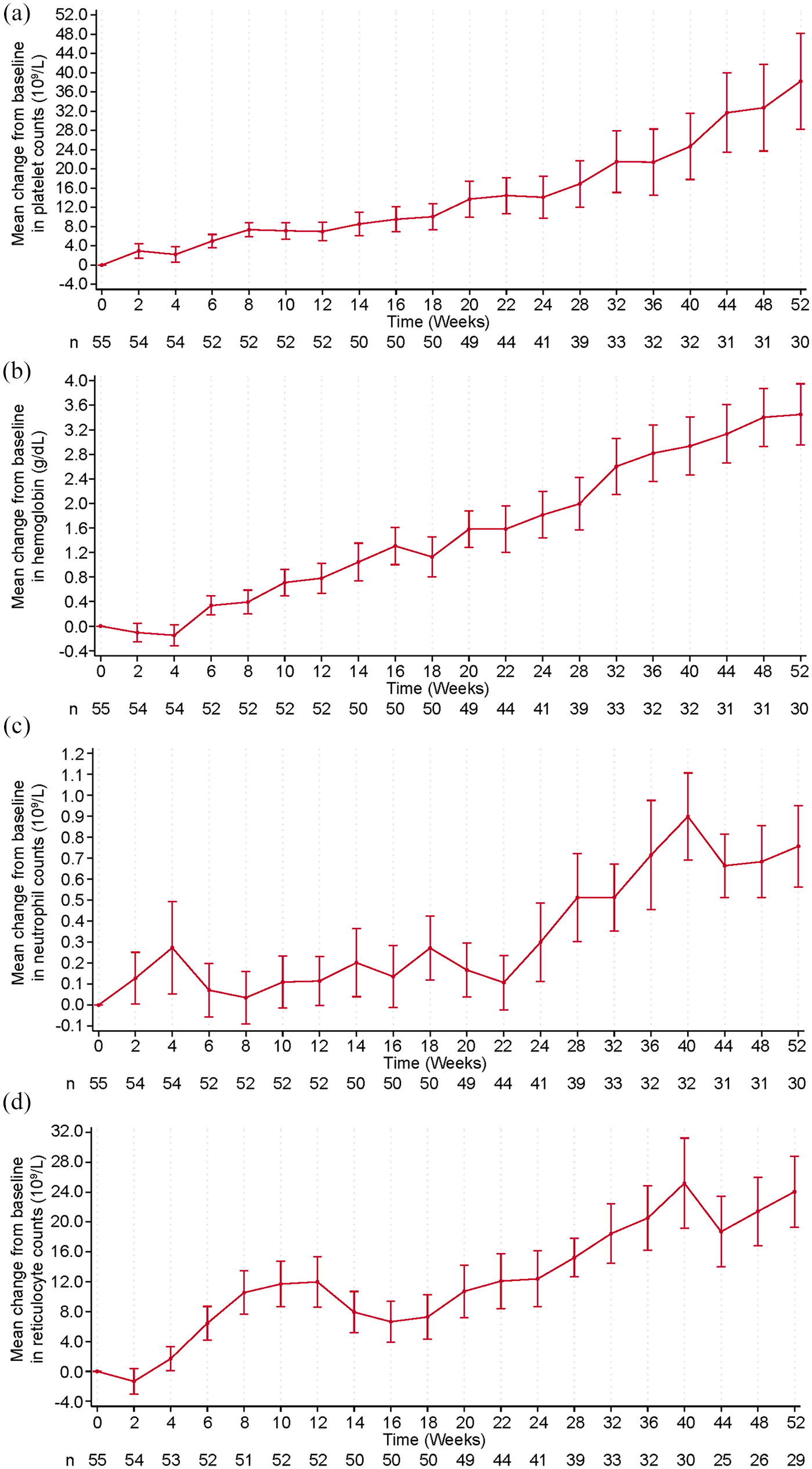
Hematologic improvements over time from baseline: Data are mean of (a) platelet counts, (b) hemoglobin concentration, (c) neutrophil counts, and (d) reticulocyte counts during treatment with hetrombopag from baseline to week 52.
Safety
Adverse events were reported in 54 (98.2%) of the 55 patients, with the most common ones being upper respiratory tract infection (22, 40.0%), increased aspartate aminotransferase (12, 21.8%), and pyrexia (12, 21.8%) (Table 3). Twenty-eight (50.9%) patients had treatment-related adverse events. The most common treatment-related adverse events were increased gamma-glutamyl transferase (7, 12.7%), increased alanine aminotransferase (ALT; 7, 12.7%), and increased aspartate aminotransferase (7, 12.7%) (Supplemental Table S3). Adverse events of grade 3 or higher occurred in 17 (30.9%) patients, with the most common ones being pneumonia (6, 10.9%), upper respiratory tract infection (3, 5.5%), and increased gamma-glutamyl transferase (3, 5.5%). Only one (1.8%) case of grade 3 increased gamma-glutamyl transferase was related to hetrombopag.
Adverse events.

Data are n (%).
Three (5.5%) of the 55 patients underwent dose reduction or treatment interruption, but none of them was considered to be drug-related (Supplemental Table S4). Three (5.5%) of the 55 patients discontinued treatment due to adverse events, including pneumonia, brain herniation, and myelodysplastic syndrome (MDS), which were not related to hetrombopag treatment according to the investigator’s judgment. Serious adverse events were reported in 15 (27.3%) patients (Supplemental Table S5), with pneumonia (5, 9.1%) being the most common; however, only one (1.8%; constipation) was related to treatment. Three (5.5%) deaths were reported. The cause of death included cardiac failure, infection, sepsis, liver abscess, and brain herniation, which were judged as not related to hetrombopag.
Clonal cytogenetic evolution occurred in two (3.6%) patients after 18 weeks of hetrombopag administration: one patient had a loss of chromosome 7 (45, XY, –7[16] XY[4]) and one had an increase of chromosome 8 (47, XX, + 8[3] XX[9]). No bone marrow fibrosis occurred. Abnormal hepatic function was an adverse event of special interest specified in the trial protocol. Abnormal hepatic function [ALT >3× upper limit of normal (ULN) and bilirubin >2× ULN], an adverse event of special interest, was reported in one (1.8%) patient on day 168 after initiation of treatment; this event was grade 2 and considered as hetrombopag-related. The elevated ALT level returned to normal on day 177 without discontinuation of hetrombopag treatment.
Discussion
In this study, we assessed the efficacy and safety of hetrombopag, a TPO-R agonist, in patients with SAA who were refractory to prior IST. The study met its primary endpoint, achieving a hematologic response rate of 41.8% (95% CI = 28.7–55.9) at week 18, which compared well across studies; for instance, a hematologic response rate of 40% (17/43) at 16 weeks was previously observed for eltrombopag. 8 There were 6/55 (10.9%) trilineage responders observed at 18 weeks for hetrombopag compared with 1/43 (2.5%) reported for eltrombopag at 16 weeks. The median time to first response was 7.9 weeks for hetrombopag compared with 12 weeks for eltrombopag.7,8 However, to date, there are no direct head-to-head comparisons of hetrombopag and eltrombopag for the treatment of refractory aplastic anemia. Comparative efficacy and tolerability trials would be of great interest. In addition, although a higher response rate (84%) was observed in a romiplostim study compared with hetrombopag or eltrombopag studies, it may be due to the different response criteria and only some patients with prior history of treatment with ATG. 9
The hematological response rate of hetrombopag at weeks 18, 24, and 52 was 41.8%, 43.6%, and 49.1%. The higher response rates with longer treatment times are generally in line with two real-world studies of eltrombopag in Europe (46% and 62%).25,26 In our study, among the 23 patients who had hematologic responses at week 18, 20 patients maintained their response through week 24, indicating the persistent efficacy of hetrombopag. In addition, we observed new responses appear with the prolongation of treatment time. Four patients who did not have hematologic response at week 18 obtained hematologic response at week 24, and four patients who did not show response at neither week 18 nor 24 achieved stable response after week 24. The number of patients with trilineage response increased from 6 (10.9%) at week 18 to 9 (16.4%) at week 24, and to 12 (21.8%) at week 52, further indicating the improvement of hematological status with the prolongation of treatment time. Among the 32 patients achieving hematologic response in week 18, 24, or 52, the mean proportion of CD34+ hematopoietic stem cells was 12% ± 15% at baseline, and then increased to 21% ± 27% after 18 weeks of treatment and 19% ± 23% after 52 weeks of treatment. Although the increase in CD34+ proportions after 18 and 52 weeks of treatment was not statistically significant compared with baseline, this result showed that bone marrow hematopoietic function was improved after hetrombopag treatment.
We observed a relatively low transfusion/injection-independent rate (Table 2), because we adopted the following strict criteria: platelet or red blood cell transfusion independence was defined as platelet ⩾20 × 109/l or hemoglobin ⩾8 g/dl for 8 consecutive weeks without platelet or red blood cell transfusion; G-CSF injection-independence was defined as neutrophil count ⩾0.5 × 109/l for 2 consecutive weeks without injection of G-CSF.
We found that more patients achieved an erythroid response than the other two lineages at week 18, but the platelet count increased more compared with baseline than the hemoglobin or neutrophil count at week 18 [platelet, 102.95 (183.89)%; hemoglobin, 19.18 (35.84)%; and neutrophil, 66.54 (161.47)%]. In addition, two patients required a reduced treatment dose, as their platelet count increased above the lower limit of dose adjustment criteria; no patients discontinued treatment in case of sustained hematological response. Of these, the platelet level of one patient increased from 20 × 109/l at baseline to 215 × 109/l at week 48, so the daily dose was gradually reduced by 2.5 mg every 2 weeks until a maintenance dose of 5 mg was reached when the patient was out of the study. The platelet level of the other patient increased from 13 × 109/l at baseline to 215 × 109/l at week 36; therefore, the dosage was reduced from 15 to 12.5 mg, and finally maintained at 10 mg.
Hetrombopag was well tolerated and its safety profile was manageable. Adverse events were reported in 98.2% of patients, and most of the adverse events (67.3%) were grades 1–2, which were transient and could be relieved after hetrombopag interruption or symptomatic treatment. Serious adverse events were reported in 15 (27.3%) patients, but most of them were associated with hemorrhage or infection caused by SAA itself, and only one event, a patient with constipation, was judged to be related to hetrombopag. Hepatotoxicity had been noted with the use of TPO-R agonists in patients with SAA;8,27,28 for instance, increased transaminases occurred in 12% of patients receiving eltrombopag therapy. 29 In this hetrombopag study, treatment-related increase in transaminase levels was observed in 8 (14.5%) patients, and most of them were grades 1–2. After liver protection treatment, transaminase levels ultimately returned to normal and did not lead to dose reduction, treatment interruption, or discontinuation. Although hepatotoxicity was commonly asymptomatic and could be controlled through appropriate treatment, monitoring liver parameters and necessary dosage modification are recommended to prevent liver injury induced by hetrombopag.
Clonal evolution (defined as new onset of chromosomal abnormalities or MDS/acute myelogenous leukemia) is a theoretical concern with the use of TPO-R agonists in SAA,8,27,28 and has been reported in studies of eltrombopag treatment for SAA, with an incidence of 18.6% after 3–13 months of treatment, 8 and most of which were abnormalities in chromosome 7. In this study, we observed two cases of chromosomal abnormalities. One patient developed abnormalities in chromosome 7 after 18 weeks of treatment and was eventually diagnosed with MDS. Another patient developed abnormalities in chromosome 8 after 18 weeks of treatment and died before an MDS diagnosis was confirmed. Since no carcinogenicity of hetrombopag was observed in mice (data on file, Hengrui) and patients with SAA have a 10–15% baseline probability of developing clonal evolution, 30 the association of clonal evolution with hetrombopag remains unclear. Further studies of hetrombopag should be conducted with very close monitoring for clonal abnormality in SAA.
Another adverse event of particular concern associated with eltrombopag is cataract, which was observed in rodent toxicological studies. 29 In this hetrombopag study, only one patient who had a history of bilateral lens cortical opacification developed bilateral lens cortical and core opacities at week 18 of treatment, which were considered as hetrombopag-related. Considering that cataracts had not been observed in preclinical studies of hetrombopag, further investigations of cataracts in studies of hetrombopag in patients with SAA are warranted.
This study has several limitations. First, this was a single-arm trial and no control group was included. Second, all patients enrolled in this study had received one course of IST before enrollment, so the efficacy data of the hetrombopag in patients who had received two or more courses of previous IST could not be obtained.
In conclusion, hetrombopag showed encouraging activity with multilineage hematologic responses in patients with SAA who were refractory to prior ATG-globulin-based IST. Hetrombopag was generally well tolerated and safe for long-term use.
Supplemental Material
sj-docx-1-tah-10.1177_20406207221085197 – Supplemental material for A multicenter phase II study on the efficacy and safety of hetrombopag in patients with severe aplastic anemia refractory to immunosuppressive therapy
Supplemental material, sj-docx-1-tah-10.1177_20406207221085197 for A multicenter phase II study on the efficacy and safety of hetrombopag in patients with severe aplastic anemia refractory to immunosuppressive therapy by Guangxin Peng, Guangsheng He, Hong Chang, Sujun Gao, Xinjian Liu, Tong Chen, Pei Li, Bing Han, Miao Miao, Zheng Ge, Xiaoyan Ge, Fei Li, Yingmei Li, Shunqing Wang, Yi Wang, Yaqi Shen, Tao Zhang, Jianjun Zou and Fengkui Zhang in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
We thank the patients and their families, and acknowledge the contributions of all investigators in this trial. We would also like to acknowledge Tengfei Zhang (PhD, Medical Writer, Jiangsu Hengrui Pharmaceuticals Co., Ltd.) for medical writing support according to Good Publication Practice Guidelines.
Author contributions
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Yaqi Shen, Tao Zhang, and Jianjun Zou are employees of Jiangsu Hengrui Pharmaceuticals Co., Ltd. Other co-authors declare no competing interests.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Jiangsu Hengrui Pharmaceuticals Co., Ltd. The authors and sponsor collaborated in data collection, analysis, and interpretation, and guaranteed the authenticity and integrity of the data. All authors had full access to the data in this study and had final responsibility for the decision to submit for publication.
Ethical approval and consent to participate
The trial was conducted in accordance with Declaration of Helsinki and the Good Clinical Practice Guidelines. The protocol and all amendments were approved by the institutional review board or independent ethics committee of each participating clinical center. All patients provided written informed consent.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.