
Abstract
Sickle cell disease, despite its recognition as a severely debilitating genetic condition affecting hundreds of thousands of neonates throughout the world each year, was not a target of pharmaceutical research focus for most of its 100-year existence in the medical consciousness. This has changed in recent years as many novel therapeutics are currently under investigation, with three new disease-modifying drugs achieving FDA approval in the last 4 years. One of these drugs, voxelotor, is especially encouraging as an inhibitor of sickling for its ability to safely improve the chronic hemolytic anemia of sickle cell disease. This was demonstrated during all clinical phases of investigation by an average improvement in hemoglobin of greater than 1 g/dL, as well as statistically significant improvements in established markers of hemolysis. While anemia itself represents a potential cause of morbidity, it is more importantly a marker of the hemolysis known to cause the long-term vascular and organ damage that makes sickle cell disease so debilitating and frequently fatal early in life. Given the recency of the approval, there has not been sufficient long-term follow-up to demonstrate improvement in the chronic sequelae of sickle cell disease as a result of voxelotor-induced improvements in hemolytic anemia. There is hope, however, based on the experience with hydroxyurea improving morbidity and mortality via reductions in sickling and improved rheology, that voxelotor may have similar long-term benefits by positively manipulating the kinetics of hemoglobin polymerization. This review aims to summarize the targeted pathobiology of sickle cell disease, the mechanism of action of voxelotor, and the safety and efficacy data from preclinical to late clinical stage investigations of this long-awaited medication, in the hopes of better informing the decision-making process behind prescribing or not prescribing it for patients in need of intervention.
Sickle cell disease: a history of neglect
It has now been 10 years since sickle cell disease (SCD) reached the dubious 100th anniversary of its first official description by James Herrick and Ernest Irons in 1910. 1 Yet despite its centenarian status in the medical consciousness, it is only in the last 5 years that technology and disease awareness have advanced enough to result in a treatment specifically designed and approved for SCD.2–5 For the first time in that long history, providers have multiple medications at their disposal targeted intentionally at the complex underlying pathobiology behind the devastating complications from which patients with SCD suffer. While disease-modifying therapy has long been available for so many other disease states, the recent FDA approval of the second, third, and fourth medications for SCD in rapid succession, over 20 years since the approval of the first in hydroxyurea, signaled entry into a radically different treatment landscape of SCD management. Patients who refuse or do not tolerate hydroxyurea are no longer limited to purely palliative symptom control, but instead have multiple options available depending on their unique disease phenotype and treatment goals. Unfortunately, despite the longstanding knowledge that hydroxyurea reduces the frequency of painful episodes, acute chest syndrome, and transfusions, and ultimately improves survival, prescribing and uptake of the drug continue to fall woefully short of the recommended universal treatment of patients with hemoglobin SS type SCD and sickle beta zero thalassemia, as well as select patients with other genotypes depending on clinical course.6,7 Symptom palliation has thus remained the predominant treatment. The availability of additional therapeutic options (assuming ongoing positive data) will undoubtedly improve the quality of life for patients with SCD, either via alternative monotherapies or adjunctive treatments to hydroxyurea. Detailed understanding regarding the history, mechanisms, efficacy, and risks of these medications (hydroxyurea, l-glutamine, crizanlizumab, and voxelotor) is necessary to shift the thinking among providers and patients alike from palliation to prevention. This review aims to provide that background specifically for voxelotor.
Treatment targeting only the downstream acute and chronic damage associated with SCD is not only costly to the healthcare system at a collective annual expense of well over $1 billion, but more importantly becomes exhausting and debilitating both in terms of medical as well as psychosocial functioning, as one sequela can often contribute to the development and worsening of another.2,8,9 The more chronic sequelae of the disease have become even more prevalent with improved survival as SCD has shifted from almost exclusively a disease of childhood to a chronic disease of adulthood. 10 For the modern patient with SCD, however, prolonged survival is often characterized by a gradual decline in quality of life secondary to accumulated multi-organ dysfunction. There has therefore remained a significant need for additional disease-modifying therapies that interrupt the deterioration of organ function that are effective, well tolerated, and easily adhered to. In November 2019, one such medication, voxelotor, was granted accelerated FDA approval for the treatment of SCD as a first-in-class polymerization inhibitor capable of improving anemia in affected patients (anemia chosen as a surrogate endpoint predicting clinical benefit for the purposes of the accelerated approval process). Although it was granted approval based on a hemoglobin increase of only 1 g/dL, many study participants achieved larger increases, and the demonstrated reduction in hemolysis leading to these increases was expected to more significantly impact patients than the hemoglobin value itself based on what is known about the pathophysiology of SCD, described below. Increasing hemoglobin by 1 g/dL is a surrogate endpoint, and clinical benefit will require verification through clinical trials that are ongoing.
Anemia in sickle cell disease
The most common sequela of SCD, and one which is not only a debilitating complication itself but also a marker of the underlying process behind much of the pathophysiology of SCD, is anemia. The chronic hemolytic anemia of SCD is more of an indirect than a direct cause of morbidity even for those patients functioning at severely low baseline hemoglobin levels (commonly as low or even less than 7 g/dL). While clinical intuition supported by experience with other conditions of chronic anemia (cancer, iron deficiency anemia, etc.) may naturally ascribe the persistent fatigue of SCD to such low oxygen-carrying capacity, the evidence is equivocal as to a direct correlation between anemia and fatigue.11–13 Outside of this possibly related symptom and the more imminent risk of severe acute anemia during aplastic crisis or hyper-hemolysis, chronic anemia may on the surface seem to be a relatively innocuous component of SCD when viewed purely as a symptom of the disease. However, with a shift in focus from the “anemia” to the “hemolysis” of chronic hemolytic anemia of SCD, it is more accurately categorized as a very important biomarker for the pathobiological processes at the heart of the disease.
In particular, hemolysis is established as a primary driver of disease severity partly as a result of studies associating anemia (as a surrogate marker of hemolysis) with the aforementioned sequelae, as well as with increased mortality.14–19 Similar associations have been found for other markers of hemolysis, in particular lactate dehydrogenase, indirect bilirubin, cell-free plasma hemoglobin, and red blood cell-derived microparticles. 15 Although the degree of chronic hemolysis varies widely from patient to patient, its essential importance to the pathobiology of SCD does not, as the endothelial activation and vascular damage caused by the release of intra-corpuscular elements drive the increased cellular adhesion, inflammation, and disturbed rheology behind the ischemia–reperfusion damage that characterizes the disease.2,15
Based on this mechanistic model for chronic organ damage, anemia has been viewed as an important surrogate marker for disease modification without waiting years or potentially decades to longitudinally assess reduction in the cumulative effect of chronic hemolysis. Prevention of hemolysis has thus emerged as an important therapeutic aim. The more upstream the therapeutic target in the pathobiological pathway, in theory the greater the potential effect on preventing hemolysis. An obvious treatment choice, therefore, outside of editing the mutated sickle gene itself, would be one that targets the most primary cause of pathology in SCD: sickle hemoglobin polymerization. Such a medication could nullify the effects of sickle hemoglobin (HbS) by forcing it to behave normally, which in turn would allow red blood cells (RBCs) to function normally in the long term and prevent or temper the lifelong onslaught of sickled RBCs on vital organs throughout the body. This is one of several beneficial effects of hydroxyurea, but as previously stated and extensively discussed in other reviews, uptake of this drug has lagged behind recommendations. 7 Additionally, despite improvement, those patients on hydroxyurea do continue to experience complications from SCD, and the achievement of a safe additive benefit via multi-agent dosing would be ideal. One promising potential target for such a medication has been recognized for decades, though it is only recently that technology has made the trial-and-error process efficient enough to make its development possible. 20
Targetable pathobiology
Hemoglobin structure
Each RBC in the human body normally contains (along with little else) approximately 300 million molecules of hemoglobin that freely diffuse through the plasma in which they are dissolved, allowing easy and very rapid offloading of oxygen during the cells’ brief traverse through terminal capillaries in peripheral body tissues.21,22 Each molecule of normal adult hemoglobin (HbA) is composed of two alpha and two beta polypeptide chains, organized as two dimers (α1β1, α2β2) associated closely in a tetrameric superstructure. Each of these homologous globin chains incorporates a heme group composed of a protoporphyrin IX subunit and a ferrous iron capable of binding one molecule of oxygen.21,23 The unique structure of the hemoglobin molecule is such that binding of an iron atom by oxygen in one of the four subunits results in the displacement of a histidine residue connected indirectly to an integral point of attachment between the two αβ dimers, inducing a 15° rotation in the arrangement between them. This conformational change ultimately results in an increased affinity for oxygen in the next hemoglobin subunit. This cooperativity produces a three-fold increase in oxygen affinity relative to completely deoxygenated hemoglobin for a tetramer with one out of four subunits bound to oxygen, and a twenty-fold increase in affinity if three out of four binding sites are filled, resulting in very efficient uptake and offloading of oxygen as needed in the lungs and distal tissues, respectively.21,23–25 The deoxygenated (and consequently lower affinity) state is referred to as the T-state (tense), while the variably oxygenated states with progressively increasing oxygen affinity are referred to broadly as R-states (relaxed).21,24,26,27 An understanding of this unique molecular design is key to understanding the pathobiology of SCD.
Sickle hemoglobin polymerization
In HbS, a single adenine to thymine base substitution translates to a hydrophobic valine replacing a hydrophilic glutamine on the beta-globin chains at position 6. In the deoxygenated state this valine is exposed on the outer surface of hemoglobin and easily becomes buried in a hydrophobic pocket of a beta-globin chain on an adjacent hemoglobin molecule. With a sufficient concentration of HbS molecules that spend a sufficient amount of time in a deoxygenated state, this process can repeat until a long polymer of HbS molecules takes shape that deforms the RBC into the recognizable sickle shape of “sickle cells,” and also damages its cell membrane, triggering the vicious cycle of pathobiological changes behind the myriad of clinical complications of SCD.23,28
Further increasing the risk for sickling is the lower affinity of HbS for oxygen in general compared with HbA, secondary not only to the tendency toward polymerization and the subsequent structural alterations of oxygen binding sites, but also due to the elevated concentration of 2,3-diphosphoglycerate, a naturally occurring and normal biproduct of glycolysis which stabilizes the T-state to promote oxygen offloading.29–31 Under normal conditions this allows for improved oxygenation and physiologic adaptability under physical stress. In a person with SCD, under physical stress the reduced oxygen affinity of the T-state becomes a maladaptive accelerant of sickling. The delay time for initiation of polymerization is inversely and exponentially related to the hemoglobin concentration.32,33 In a homozygous HbS patient with >95% HbS that wants to remain deoxygenated and wants to polymerize, there is thus little natural resistance against the initiation of this potentially deadly process. Other hemoglobin variants, however, provide us with valuable information that has proven life saving for patients in the past, and hopefully many more in the future. Since neither HbA, fetal hemoglobin (HbF), or oxygenated HbS take part in polymerization interactions, a reduction in deoxy-HbS concentration via an increase in any one of these can alter the kinetics of polymerization and provide a sufficiently prolonged delay time for RBCs to pass through the hypoxic environment of distal capillaries and arterioles without sickling prior to reoxygenation in the lungs.32,34–37 The lack of symptoms in patients with sickle cell trait (i.e. approx. 50% HbS and 50% HbA), and the improvement in symptoms for patients with HbF levels of 30% secondary to either hydroxyurea effect or hereditary persistence of fetal hemoglobin indicate that even a relatively small dilution of HbS concentration by less than 50% can be therapeutic.6,32,34,35,38,39
The translation of polymerization to pathology
HbS polymerization causes oxidative damage to the cell membrane as well as dehydration and rigidity of the RBC. As a result of various signals of damage and plasma membrane perturbations, the cells become “sticky” and are more likely to bind leukocytes, platelets, and the endothelium of the small diameter vessels through which they are typically traveling at the time of sickling. 40 At the same time the less deformable sickle cells move less freely through these narrow passages, maintaining prolonged contact with endothelium and allowing these adhesive interactions. These factors all contribute to vaso-occlusion, hemolysis in the setting of increased shear stress, and damage to the vasculature. The tendency toward rapid hemolysis reduces the lifespan of these cells significantly, from approximately 120 days to less than 3 weeks, after which they undergo both intravascular and extravascular hemolysis. 41 Chronic hemolysis (primarily intravascular hemolysis) results in the release of toxic intracellular substances including arginase, heme, and other strong oxidizing agents that serve to directly damage the vascular endothelium, consume nitric oxide as well as the substrate for its production arginine, and activate both the inflammatory and coagulation cascades. The resulting vicious cycle causes narrowed capillaries, greater tendency to form microthrombi, and further increased adhesion between the endothelium and all three blood cell types.14,15,42,43 Given the obvious ubiquitous nature of blood in the body, the potential for this process to occur anywhere and everywhere puts patients with SCD at constant risk of acute and chronic organ and tissue damage.
The translation of pathobiology to pharmacology
Three medications have been approved in the last 5 years for the treatment of SCD, each of which has a different target within the aforementioned cascade of events from gelation (the initial clustering of hemoglobin molecules that serve as the nidus for polymerization), to the oxidative stress produced by free radicals spilling into the circulation during hemolysis, to the increased expression of adhesion molecules on activated vascular endothelium.3–5 The goal of all three is to interrupt the interplay and positive feedback between a toxic microenvironment, disturbed rheology, prolonged hypoxemia, sickling, and hemolysis. The target of voxelotor is the furthest upstream, as it is capable of interrupting sickling itself by altering the kinetics of gelation, the primary event in initiating and compounding the above process. The possibility of even slight alterations in HbS polymerization kinetics to have massive therapeutic implications has been excellently summarized by Eaton and Bunn, and served as the basis for voxelotor’s development. 44 It achieves polymerization inhibition by allosterically stabilizing the R-state of hemoglobin in much the same way that 2,3-DPG stabilizes the T-state to achieve the opposite effect. This stabilization increases the ratio of oxygenated to deoxygenated HbS even in the presence of significant hypoxia, thereby achieving the important prolongation in polymerization delay time mentioned previously. 45
Inhibition of polymerization: pathway to discovery
The potential for inhibiting sickling by pharmacologically increasing the oxygen affinity of HbS was recognized as early as the 1970s during in vitro investigations of the effect of HbS binding a number of different aromatic aldehydes. 20 Variations of benzaldehyde and vanillin in particular were shown very early on to result in a left shift of the oxygen dissociation curve of hemoglobin by bringing into association the N-terminal valines (note: distant from the disease-causing βVal6 of SCD) of each alpha globin chain in a given tetramer via a Schiff-base and a hydrogen bond, respectively.20,27,46–49 The conformational change brought about by this binding resulted in increased oxygen affinity, known now to be secondary to stabilization of the R-state of hemoglobin. The path from these discoveries in the lab to the approval of a safe and effective medication in 2019 has obviously been very long, due in part to the inefficient trial-and-error process of selecting a molecule with specificity for the preferred site on hemoglobin from among countless aromatic aldehyde candidates, some of which in fact reduced oxygen affinity.27,50 Primarily, though, and especially in the past 10–15 years as different research groups zeroed in on effective compounds, progress was limited by insufficient oral bioavailability and by poor partitioning of drug candidates into the RBC from plasma, necessitating high therapeutic doses that proved to be intolerable. 27 Recent successful in vitro candidates have bound hemoglobin with a 2:1 stoichiometry due to a separate drug molecule binding each alpha globin chain in each tetramer, thus further increasing the required dose.47,51–53 Voxelotor, formerly GBT-440, was able to overcome these challenges.
50 years later: first in class
In studies leading up to the selection of voxelotor for advancement from among a number of related candidates, it was found to have critically unique properties relative to the long lineage of previously investigated aromatic aldehydes. Consistent with those other molecules, voxelotor binds via reversible Schiff-base linkage to αVal1 on hemoglobin. Differentiating it, however, is the hydrogen bond it forms with αSer131 on the alternate alpha chain of the same hemoglobin tetramer. 53 Previous iterations of the drug, similarly to so many molecules studied over the last several decades, required a second molecule of drug to bind the alternate αVal1 on the second alpha globin chain of the tetramer in order to bridge the gap between the two polypeptide chains and maintain the R-state. The larger molecule completing this association more efficiently (owing to a bicyclic aromatic structure rather than a single aromatic ring) importantly resulted in its binding hemoglobin (and producing a functional change in oxygen affinity) with 1:1 stoichiometry rather than 2:1. 53 Furthermore, voxelotor in these preclinical studies was found to have in murine models an oral bioavailability of 60%, a T1/2 of 19.1 h., and a RBC:plasma partitioning ratio of 150, all of which were major breakthroughs relative to previous results in terms of the drug dose that would be necessary to achieve therapeutic effect while avoiding toxicity. 53
Preclinical results
Efficacy
Preclinical in vitro and ex vivo investigations of voxelotor in whole blood from patients with SCD and blood from treated mice, respectively, showed pharmacodynamic effects consistent with the above. HbS deoxygenated in vitro without pre-incubation with voxelotor was found to be 76% deoxygenated after 2 h, whereas HbS incubated stoichiometrically with voxelotor was only 17% deoxygenated. This increased oxygen affinity was demonstrated to be dependent on voxelotor concentration. Given the effect of oxyHb on polymerization kinetics previously described, such a dramatic change in affinity was expected to reduce polymerization of HbS (and in turn to reduce sickling) in a hypoxic environment, in accordance with the described mechanism of action of aromatic aldehydes and with results published previously for other related compounds. This proved to be the case, as the polymerization delay time as measured by optical density increased from 9 min to 18–22 min in the presence of 20–30% modified HbS, equivalent to the known effect of the same percentage of HbF on delay time. 45 Sickling was similarly inhibited by 30% Hb occupancy with voxelotor, as after 20 min of deoxygenation to 40 mmHg (approximately equivalent to the partial pressure of oxygen in venous blood in vivo) there was no increase in sickle cells in voxelotor-treated samples. In contrast, there was a greater than 6-fold increase in sickling in untreated samples at the same oxygen tension. 45 As expected given the primacy of HbS polymerization and sickling at the heart of disturbed RBC functionality and survival in SCD, these in vitro studies also found prolonged RBC half-life (indicating reduced hemolysis), improved deformability, and decreased whole blood viscosity in deoxygenated samples incubated with voxelotor.45,54–56 Impressively, viscosity of deoxygenated, voxelotor-treated blood improved not only compared with untreated deoxygenated blood, but also compared with untreated oxygenated blood. These results suggested significant potential of increasing oxygen affinity to inhibit downstream effects, and potentially (via improved rheology) to limit the time spent by RBCs in the maximally hypoxic environment of the narrowest terminal capillaries in the body.
Early safety concerns
Despite the demonstrated effects of voxelotor in preclinical studies of reduced sickling and improved rheology, and the anticipated benefits to which this would translate in vivo, there remained concerns about the safety of its entire class of medication. These concerns still remain for many despite published safety data from clinical trials, due to the rapid approval with what some deem to be inadequate long-term data. First among them is the potential risk for reduced tissue oxygen delivery due to too severe a left shift in the oxygen dissociation curve. The very efficient cooperative binding and releasing of oxygen by hemoglobin depends on oxygen affinity being strong enough to bind oxygen at its high partial pressure in the capillaries of the lung, but weak enough to release oxygen at the lower partial pressure of active tissues.
The concern with voxelotor, which was initially appropriate given experience with some previously studied aromatic aldehydes inducing too great an oxygen affinity to be clinically useful, was that while sickling may have been inhibited by a percentage of hemoglobin molecules binding voxelotor, oxygen extraction in tissues would also be inhibited in those modified hemoglobin molecules. During the drug’s development when preclinical data from voxelotor analogs indicated the p50 for modified hemoglobin would be shifted as far or even farther than that of HbF, some asked the important question: for a patient population that already has reduced tissue oxygen delivery due to severe anemia, especially in the setting of exertion or acute respiratory failure, would a targeted 30% modification reduce oxygen delivery by an additional 30%? 57 A real reduction of this magnitude in functional oxygen content, in a person already at very high risk of stroke secondary to sickle cell-related cerebrovascular disease, could increase the risk of stroke dramatically. 58
Preclinical testing of voxelotor itself in whole blood from patients with SCD, however, indicated that the p50 was not left shifted as much as feared, and in fact only normalized a typically right shifted p50 of SS hemoglobin (32 mmHg) to around the p50 of normal AA hemoglobin (28.6 mmHg) (Figure 1). Additionally, voxelotor-modified hemoglobin was shown to still respond to the natural right-shifting substances (promoting oxygen offloading) concentrated in metabolically active tissue: H+ and 2,3-DPG. 59 Some have expressed concern, though, that even slightly decreased oxygen offloading in the brain would, via normally functioning metabolic sensors in the brain, increase cerebral blood flow, which in the presence of chronic cerebrovascular disease could again result in stroke. Even in wild-type mouse models with presumed normal baseline p50 (i.e. not right shifted as in SCD), data obtained via pimonidazole staining and sectioning indicated there was no reduced oxygenation in any organ studied, including the brain. Additionally, treated mice had significantly improved tolerance of hypoxia to 5% of normal over a 2 h period, with 83% of treated mice surviving 2 h of hypoxia compared with 0 untreated mice surviving the same.60–62

Oxygen dissociation curves for HbSS before and after treatment with voxelotor (Panel A) indicate only a modest left shift to achieve an overall oxygen affinity similar to that of normal Hb and less than that of HbF (Panel B). The targeted Hb occupancy by voxelotor at the approved dose, as evidenced by the above and similar data, is not sufficient to cause dangerous reductions in oxygen delivery to tissues where the pO2 is physiologically reduced.
Although the significant (>80%) reduction in p50 for voxelotor-modified HbS is a valid concern, it is a concern that has long been recognized for this drug class and was addressed both in those same preclinical studies as well as in clinical trials by the inclusion of erythropoietin (EPO) as a biomarker of oxygen delivery, with the thought that an elevation in EPO in voxelotor-treated patients would indicate some degree of reduced oxygenation that would warrant significant concern. Both phases of drug development have been reassuring from this standpoint. Cerebral blood flow has also been studied using magnetic resonance imaging (MRI) as part of a secondary study within the phase III clinical trial of voxelotor (described below). 64
A second concern has been raised regarding a potential increase in viscosity secondary to an increase in hematocrit. This concern largely stems from transfusion practices for SCD and the long-recognized risk of “over-transfusing” to a hemoglobin much higher than 10 g/dL. 65 Relative elevations in hemoglobin/hematocrit have a demonstrated association with those sequelae of SCD traditionally thought of as more of the vaso-occlusive complications (vaso-occlusive pain crisis, acute chest syndrome, and osteonecrosis).42,66,67 During the development of voxelotor, this was thought to be less of a concern due to what would presumably be a pan-cellular distribution of the drug and thus reduced sickling/improved deformability in all (or nearly all) RBCs. This differs from the effect of transfusions in diluting the overall percentage of sickle cells with normal RBCS to reduce the risk of vaso-occlusion. The risk in that scenario is related to the ongoing presence of sickling and the compounding effect on viscosity of a suddenly elevated hematocrit. Even hydroxyurea, which does target polymerization and limit sickling via the introduction of HbF molecules into the intracellular milieu, does not have a uniform effect in all RBCs, resulting in some F cells with low sickling potential and others with low HbF and persistent high sickling potential.35,68
A related concern surrounding viscosity and altered rheology is the risk of complications if patients were to suddenly stop the medication. Experience again with chronic transfusions for reduction of stroke risk indicates that sudden cessation of transfusion programs rapidly results in a dramatically increased risk of stroke in the months immediately following cessation.69,70 This represents a slightly different set of circumstances for voxelotor than the above. The goal of voxelotor is to increase hemoglobin via a reduction in hemolysis, but since the binding of the drug to HbS is rapidly reversible, sudden cessation of the medication would lead to a return of typical sickle cell pathobiology with an atypically greater hematocrit that could theoretically cause increased complications from increased viscosity. The counter to this argument can be gleaned from data from a sub-analysis of the STOP2 trial, which focused on the effect of hemoglobin level on risk of stroke following cessation of transfusions as evidenced by transcranial Doppler (TCD) values. The study found that following cessation of transfusions there was a negative correlation between TCD velocity and hemoglobin. 71 Although the comparison is not apples to apples in terms of the cause or degree of hemoglobin elevation between a STOP2 patient and a voxelotor-treated patient, the data are reassuring. This concern was further addressed during clinical trials of voxelotor, with evidence to follow. To date, there have been no reports of serious adverse events in patients who have discontinued the medication abruptly, including in the phase I/II trial that followed patients for 30 days from final drug dose, long enough in theory for a return to near baseline hemoglobin and RBC health status given the 10–20 day lifespan of sickle cells. Nonetheless, safety monitoring of this new drug class was (and continues to be) an important part of characterizing its utility, with this issue of sudden cessation perhaps chief among the potential concerns.
Clinical trials
Phase I/II
In the phase I/II trial of voxelotor basic safety and tolerability were addressed, as well as what turned out to be a positive translation of the aforementioned in vitro and ex vivo results into clinical benefit for patients. The randomized, double-blind, placebo-controlled study evaluated pharmacokinetic/pharmacodynamic (PK/PD) characteristics of voxelotor as well as its safety in healthy volunteers and patients with SCD. 72 With an eye toward the potential risks described above regarding functional oxygen content on the one hand and hyperviscosity on the other hand, those with hemoglobin levels <6.0 g/dL or >10.4 g/dL were excluded, in addition to those with recent transfusions or hospitalizations. Additionally, there was a predetermined plan in the protocol to dose reduce any participant who had an increase in hemoglobin of >2.0 g/dL over baseline (one patient did require a reduction at day 15 for this reason). Of note, results from the later phase III trial were reassuring regarding this concern, as many participants achieved Hb increases of >2 g/dL without evidence of increased viscosity-related adverse events (AEs). The study enrolled participants in succession into multiple placebo-controlled dosage cohorts: 500 mg/day, 700 mg/day, and 1000 mg/day. All dosages were taken for 28 days, and following safety review two additional 90-day cohorts were enrolled at doses of 700 mg/day and 900 mg/day in succession. Participants in the 900 mg/day cohort were then offered enrollment in an open-label extension study for 6 months. 72
There were no grade 3 or greater treatment-emergent adverse events (TEAEs) in any cohort, and the most common grade 1 or 2 TEAEs were headache and pain, with the majority of serious AEs being grade 3 vaso-occlusive crisis (VOC) occurring off treatment and felt to be unrelated to voxelotor. All VOCs occurred during post-treatment follow-up except one which occurred following a dose hold and reduction, but did not occur in close temporal proximity to drug discontinuation so were unlikely to result from the aforementioned “sudden cessation” effect. One participant did discontinue treatment due to rash, and another required a temporary dose reduction for the same. Three other dose reductions were for nausea (1) and elevated hepatic enzymes (2). Overall, however, voxelotor was well tolerated, as participants were compliant with taking 91% of planned doses, with none missing more than four doses. Again consistent with the preclinical data that made voxelotor such an attractive candidate relative to other tested molecules, voxelotor induced dose-dependent increases in hemoglobin oxygen affinity indicating excellent bioavailability, a terminal half-life of 1.5–3 days supporting once-daily dosing protocols, and a favorable RBC:plasma partitioning of 60–90:1, theoretically minimizing the chances of off-target toxicity.63,73
Concerns about increased oxygen affinity impairing oxygen extraction were also addressed and treatment during the investigation did not result in a concerning safety profile. Participants in the 90-day cohort underwent cardiopulmonary exercise testing at baseline, day 28, and day 91. There was a smaller reduction in peak exercise VO2 max in the pooled treatment group than in the placebo group (−0.4 mL/min/kg versus −2.4 mL/min/kg) although the difference was not statistically significant. As a secondary measure of tissue hypoxia that was feared to potentially result from increased hemoglobin oxygen affinity, EPO levels were monitored and actually were lower on day 90 for both the pooled treatment group and the placebo group, with no significant difference between the two, indicating no change in oxygen delivery to tissues.
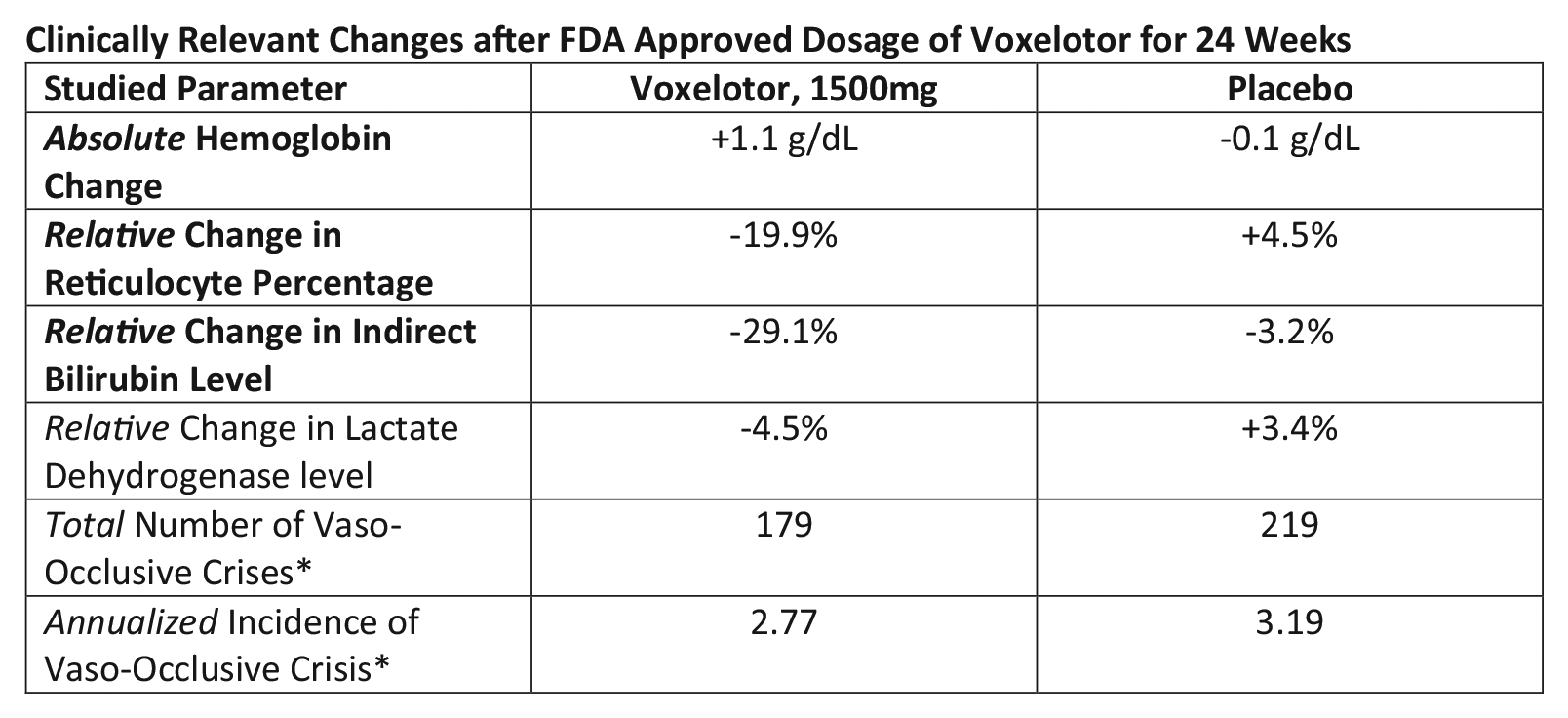
In terms of the efficacy of voxelotor in improving RBC parameters and anemia, the phase I/II trial provided evidence consistent with preclinical trials that was sufficient to support a phase III efficacy trial. There was improvement in hemoglobin levels and markers of hemolysis by 2 weeks of treatment duration. Long-term dosing for at least 90 days resulted in statistically significant improvements in hemoglobin (+1 g/dL), reductions in unconjugated bilirubin and reticulocyte percentage as markers of hemolysis, as well as a reduction in the percentage of sickled RBCs (−74% versus +6.9% for placebo). 72
Phase III
The phase III randomized clinical trial enrolled participants in a balanced ratio to receive either placebo, voxelotor 900 mg daily, or voxelotor 1500 mg daily for up to 72 weeks, with the primary endpoint measured at week 24. Those on chronic transfusions or with recent transfusions were again excluded. 4 Hydroxyurea use was not an exclusion criterion. Some 16% of participants randomized to receive voxelotor and 18% of those in the placebo group fell in the age range 12–18 years, and the rest of the participants in both arms were >18. In total, 90 participants were randomized to receive 1500 mg voxelotor, 92 to receive 900 mg, and 92 to receive placebo, and median duration of treatment for each of the three groups at study completion was between 36 and 43 weeks. Some 51% of participants in the 1500 mg group and 33% in the 900 mg group had a >1 g/dL increase in hemoglobin at week 24 compared with 7% in the placebo group in the intention-to-treat analysis. The mean changes in hemoglobin were +1.1 g/dL, +0.6 g/dL, and −0.1 g/dL in the 1500 mg, 900 mg, and placebo groups, respectively, with a small number of participants in both treatment groups achieving hemoglobin increases of >3 g/dL (Figure 2). At 72 weeks of treatment these improvements persisted, with about 87% of participants achieving a >1 g/dL improvement, 59% achieving a >2 g/dL improvement, and 20% a >3 g/dL improvement. These increases largely appeared by 2 weeks of treatment and levels remained stable subsequently and were irrespective of concurrent hydroxyurea use. With an eye toward the hemolysis that is really the focus of interest in therapeutic potential of the drug, there was again a statistically significant reduction in indirect bilirubin as well as reticulocyte percentage at week 24 for those in the 1500 mg group that persisted at week 72.4,74 Although patient-reported outcomes have as yet been lacking as an endpoint assessing efficacy, a datapoint that will be very important moving forward, the investigators did analyze data from the Clinical Global Impression of Change, a provider-reported outcome measure assessing overall expert impression of clinical status. 75 Providers were blinded to both intervention and changes in lab parameters when completing the assessment at weeks 24 and 72. There was a statistically significantly greater number of participants rated as “very much improved” or “moderately improved” in the 1500 mg voxelotor group compared with the placebo group (74% versus 47%, p < 0.01) at 72 weeks. 75
Statistically significant changes in
In additional clinically relevant analysis, there was a trend toward lower frequency of VOC in the treatment groups compared with placebo, though the study was not powered to detect a statistically significant change in this clinical endpoint. A lack of early increase in VOCs and the recently reported improvement in VOC frequency with increasing hemoglobin are again reassuring in terms of the aforementioned concerns related to the association between higher hemoglobin and viscosity-related vaso-occlusion. 76 In fact, although there was not a statistically significant overall reduction in VOC frequency associated with voxelotor treatment, the lowest rate of VOC was found in those achieving the highest hemoglobin levels (>12 g/dL). This inverse relationship can reasonably be explained by the improvements in overall rheology counteracting the effects on viscosity of higher hematocrits found in prior studies of patients with high levels of unmodified HbS.
Additional safety data
In terms of safety, there was no difference in the frequency of AEs between groups (SCD related or treatment related), with the most common being headache and diarrhea. There was again no rise in EPO level as a result of voxelotor treatment, and in fact in the 1500 mg group there was a trend toward a reduction in this marker of tissue oxygen delivery (Figure 3). Additionally, the previously described concerns related to cerebral oxygenation and perfusion, which have been lessened by the preclinical data in mouse models, were further ameliorated by an ancillary study to the HOPE Kids 1 study, preliminary data for which has been published for three pediatric participants. 64 These three participants underwent MRI/magnetic resonance angiography at baseline and following treatment with voxelotor, and were shown to not only have no increase in cerebral perfusion to suggest reduced oxygen offloading in the brain, but two of the three actually had decreased cerebral perfusion, indicating an improvement in oxygen delivery. These data not only support (albeit with limited numbers) the safety of voxelotor in terms of cerebral perfusion, but an additional benefit for the pediatric age group, for whom cerebral blood flow velocity as measured by TCD represents a vitally important risk factor for stroke. While voxelotor is currently only approved for patients aged 12 and up, the HOPE Kids 1 trial is ongoing with an ultimate goal of demonstrating safety and efficacy in a stepwise fashion for younger and younger patients to a minimum age of 9 months. 77 The early returns are that voxelotor safely improves RBC health and reduces hemolysis in similar fashion to its effect in adults, but with higher PK exposure in those younger than 12 years old, indicating the need for reduced weight-based dosing in this population.78,79 The effect of the drug on TCD velocities for children as young as 2 years old is currently being investigated in the HOPE Kids 2 trial. 80
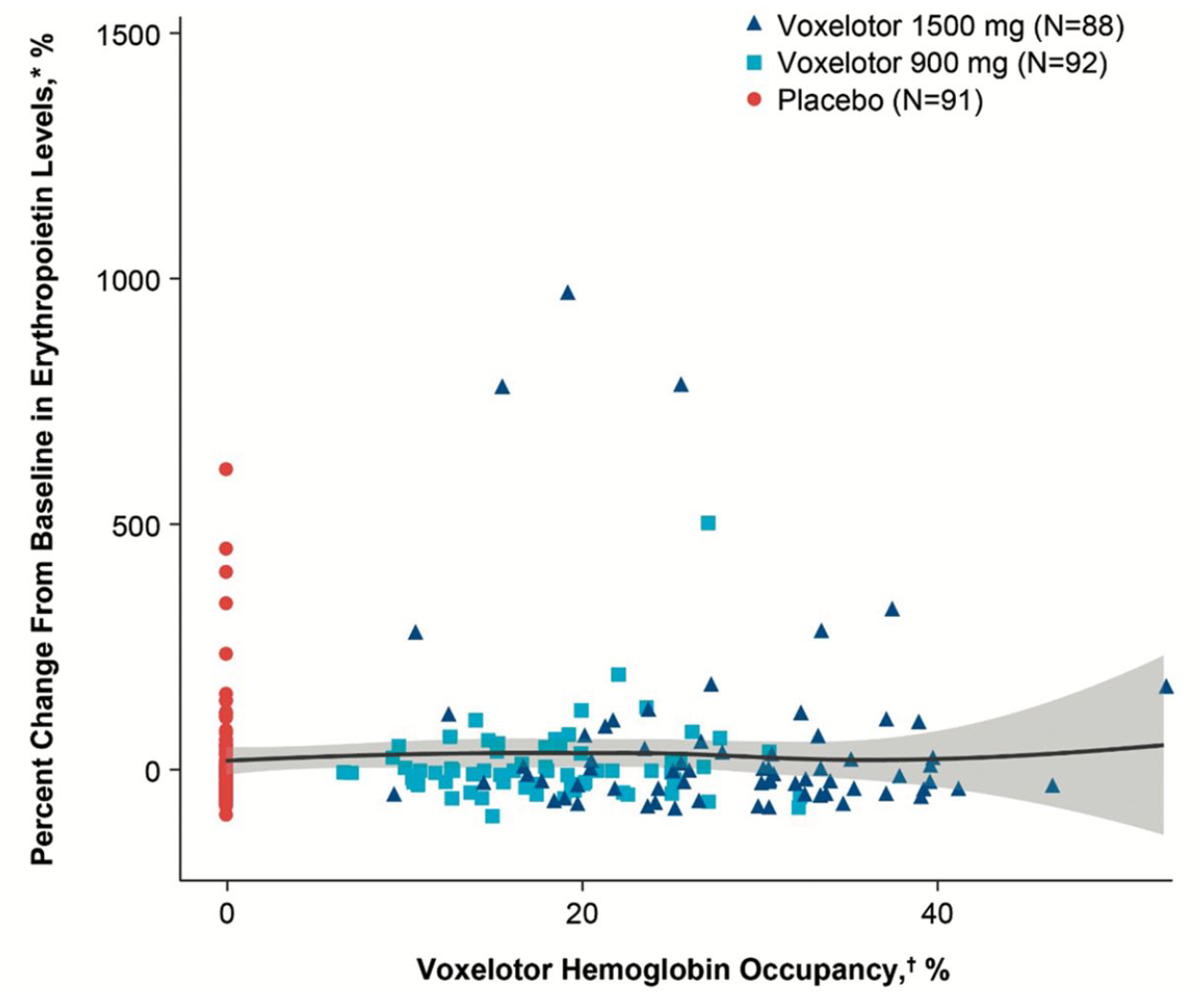
Change in erythropoietin (EPO) levels after 24 weeks of treatment with voxelotor, compared with placebo. The lack of increase in EPO indicates adequate tissue oxygenation despite increased Hb oxygen affinity, as hypoxia in the kidney especially stimulates EPO production. There was a nonsignificant trend toward reduced EPO at the FDA-approved dose of 1500 mg, suggesting oxygenation may in fact be improved by voxelotor treatment.
Separate from the clinical trials described, a case series was published detailing the courses of seven patients with severe SCD treated with voxelotor on a compassionate use basis. Five of the seven patients required frequent transfusions and one was refractory to transfusions, and four patients had baseline oxygen saturations of <95% with two on chronic supplemental oxygen. Two patients died of longstanding complications of their disease, deemed unrelated to voxelotor treatment. Five out of seven patients had rapid increases in hemoglobin of greater than 1 g/dL, with the most significant increase being 5.4 g/dL. Transfusion requirements decreased 60% on voxelotor, and two patients who required frequent transfusions no longer required any afterward. Additionally, four patients had chronic hypoxemia with baseline oxygen saturations of <95%, and two of these required long-term supplemental oxygen. All four patients experienced improvement in oxygen saturation to >98% on voxelotor, and both patients requiring long-term oxygen were able to discontinue it. One of these two underwent a 6-minute walk test and had incremental improvement from baseline to 24 weeks of voxelotor treatment. There were similar data showing no increase in heart rate. 81 There was additionally a 67% reduction in number of admissions for VOC pain when comparing the 24-week period on voxelotor to the 24 weeks prior to drug initiation. These data argue against concerns regarding oxygen extraction throughout the body, but specifically in the heart and musculature.
These anecdotal data and the above cerebral perfusion data show no indication beyond a theoretical risk that voxelotor reduces oxygen delivery, and in fact the data seem to suggest exactly the opposite with variable significance. In light of the lack of major acute events in the first week of therapy or in the weeks immediately after drug cessation within the clinical trials, the main concerns described previously should still be considered, but should factor into risk versus benefit analysis to a much smaller degree when prescribing voxelotor for patients. Voxelotor represents a promising upstream modulator of sickling with the potential to impact care in the same way hydroxyurea began to 30 years ago. While hydroxyurea has proven time and again to very effectively improve many of the symptoms of SCD and certainly holds benefit beyond its strictly approved indication, certain sequelae such as pulmonary hypertension and stroke still occur in patients treated with the medication although it does confer some protection.6,82 Irrespective of its benefits, lack of patient buy-in has limited the realization of its potential benefits even decades after its FDA approval for this patient population. 7 A comparison cannot be made between the proven long-term benefits of hydroxyurea and the theoretical benefits of voxelotor, but voxelotor’s favorable side effect profile, once-daily dosing, and its development as purely a medication for SCD will hopefully make it more amenable both to patients who do not take hydroxyurea and those who do (equivalent safety and efficacy regardless).4,83
Conclusion
In short, in a phase III multiple-dose randomized clinical trial, voxelotor was shown to safely increase hemoglobin levels and reduce markers of hemolysis in a dose-dependent manner, with a trend toward these changes contributing to an annualized reduction in VOCs, though VOC was not a targeted endpoint of the study. With these results in mind, and the potential for as yet undetermined long-term benefit secondary to a reduction in hemolysis-induced vascular damage, it is important to appreciate, but not depend on, phase IV post-approval surveillance before deeming this a safe medication. Early returns from the mentioned compassionate use experience as well as the phase III clinical trial indicate there may be important clinical benefits (improvement in leg ulcers, improvement in oxygen requirement, improvement in hospitalizations) that have yet to be fully described given the accelerated approval. Most importantly, however, given the demonstrated improvement in hemolytic anemia and what is known about the role of hemolysis in the long-term sequelae of SCD and in increasing mortality risk, ignoring the existing safety data focused on adequate tissue oxygen delivery and demanding data from a greater number of patients prior to prescribing this very promising medication would be a disservice to the patients who have waited so long for a meaningful step forward in treating their disease. While it is important to avoid thoughtlessly prescribing a medication simply because it is new and unique, it is similarly important to recognize the faulty inertia of decades of stagnant treatment experience that may fool us into thinking the status quo is good enough. Disease-modifying therapy options are desperately needed for patients with SCD, who deserve every chance at improved quality of life. Toward this end, the evidence supports voxelotor as a very good option with a mechanism of action that is completely unique from other recently approved medications in such a way as to warrant not only unique indications for prescription, but also (based on ongoing trials) the systematic use of combination therapy in the future. Based on the available evidence, in those patients >12 years of age with hemoglobin <10 g/dL, those who are either unable or unwilling to take hydroxyurea, and in those on hydroxyurea who continue to have anemia and ongoing hemolysis, it should be considered as a safe and potentially efficacious adjunct therapy.
Footnotes
Acknowledgements
None
Author contributions
All four of the listed authors made critical contributions to the conception, design, drafting, and revision of the manuscript. All authors gave final approval of the completed work and agree to be accountable for all aspects of the work ensuring integrity and accuracy.
Conflict of interest statement
Alexander Glaros has no conflicts of interest to disclose. Reza Razvi has no conflicts of interest to disclose. Nirmish Shah: Novartis (speaker, consultant, research); Global Blood Therapeutics (speaker, consultant, research); Bluebird Bio (consultant); Alexion (speaker). Ahmar Zaidi: Global Blood Therapeutics (speaker, consultant, research funding); Emmaus Life Sciences (consultant, research funding); Cyclerion (consultant); Bluebird Bio (consultant); Chiesi (consultant); Novartis (consultant).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Ethics approval was not sought for this manuscript as it does not represent original research involving human or animal subjects, but rather is a review of previously published data and research related to a specific topic.