
Abstract
Individuals with sickle cell disease (SCD) are living further into adulthood in high-resource countries. However, despite increased quantity of life, recurrent, acute painful episodes cause significant morbidity for affected individuals. These SCD-related painful episodes, also referred to as vaso-occlusive crises (VOCs), have multifactorial causes, and they often occur as a result of multicellular aggregation and vascular adherence of red blood cells, neutrophils, and platelets, leading to recurrent and unpredictable occlusion of the microcirculation. In addition to severe pain, long-term complications of vaso-occlusion may include damage to muscle and/or bone, in addition to vital organs such as the liver, spleen, kidneys, and brain. Severe pain associated with VOCs also has a substantial detrimental impact on quality of life for individuals with SCD, and is associated with increased health care utilization, financial hardship, and impairments in education and vocation attainment. Previous treatments have targeted primarily SCD symptom management, or were broad nontargeted therapies, and include oral or parenteral hydration, analgesics (including opioids), nonsteroidal anti-inflammatory agents, and various other types of nonpharmacologic pain management strategies to treat the pain associated with VOC. With increased understanding of the pathophysiology of VOCs, there are several new potential therapies that specifically target the pathologic process of vaso-occlusion. These new therapies may reduce cell adhesion and inflammation, leading to decreased incidence of VOCs and prevention of end-organ damage. In this review, we consider the benefits and limitations of current treatments to reduce the occurrence of VOCs in individuals with SCD and the potential impact of emerging treatments on future disease management.
Introduction
Sickle cell disease (SCD) is an autosomal recessive disorder caused by mutations in the HBB gene that encodes the β-globin subunit of HbA, and is characterized by the presence of red blood cells (RBCs) that contain hemoglobin S (HbS) without additional normal hemoglobin A. 1 Affected individuals may have two copies of the HbS mutation [hbSS or sickle cell anemia (SCA)], or may have one copy of HbS and another gene that produces a different abnormal hemoglobin (such as hemoglobin C or D), or a quantitatively deficient hemoglobin with thalassemia defect. Under conditions of hypoxic stress, HbS-containing RBCs undergo shape transformation to the characteristic sickle form, become inflexible, and break down easily in the circulation. Furthermore, the deformed RBCs exhibit increased stickiness, causing abnormal adherence to the endothelium, which, in combination with activated neutrophils and platelets, increases the risk of occlusion of the microcirculation. Complications of SCD include unpredictable, recurrent acute pain, as well as significant multiorgan dysfunction and premature mortality.2,3 The impact of these complications on affected individuals’ quality of life is profound.
The hallmark of SCD is pain, necessitating recurrent emergency department (ED) and/or hospital visits.3,4 An acute vaso-occlusive crisis (VOC) is an important driver of this pain, and is a major cause of morbidity and acute care utilization in this population.5–8 However, the unpredictably of painful events, use of different pain definitions, the subjective nature of pain, and challenges differentiating acute and chronic pain highlight the limitations in assigning specific pain events to an underlying cause in SCD. The principal cause of VOCs is microvascular occlusion, leading to increased inflammation and tissue ischemia-reperfusion injury.9,10 Treatments for VOC have focused largely on the symptomatic management of the acute painful episode as opposed to prevention. However, specific therapies are needed to reduce the occurrence of these episodes and decrease the associated tissue and organ damage. Several investigational treatments are being studied that target the pathologic mechanism of the vaso-occlusive process and that have the potential to reduce the frequency and severity of VOC. A detailed description of all agents that target the vaso-occlusive patho-biologic process have been the subject of several recent reviews, including one that examines the pathophysiology and development of new agents. 11 In this review, we specifically discuss the current and emerging therapeutic options for reducing VOC in SCD.
Pathogenesis of acute VOCs: adhesion and inflammatory processes
The pathophysiology of SCD is initiated by the presence of HbS, which polymerizes under conditions of reduced oxygenation, causing deformation and damage to the RBC membrane.3,12 Hemolysis caused by unstable HbS results in release of free hemoglobin, labile iron, and oxidative stress (Figure 1).
13
This, in turn, causes inflammation and activation of neutrophils, platelets, and endothelial cells through promotion of release of placenta growth factor and endothelin I, and the activation of toll-like receptor 4 and NALP inflammasome signaling.
13
Ultimately, this leads to adhesion of RBCs, neutrophils, and platelets to the endothelium, resulting in vaso-occlusion.13,14 Free hemoglobin also directly consumes nitric oxide, leading to endothelial dysfunction.15,16 In addition, hemolysis also causes release of arginase I, which depletes plasma

Contribution of intravascular hemolysis to vasculopathy and vaso-occlusion. Intravascular hemolysis produces free hemoglobin, which drives Fenton reactions to produce oxidants and scavenges NO by a deoxygenation reaction. Intravascular hemolysis also releases red cell arginase 1 into plasma, where it can deplete plasma
Thus, the processes underlying VOC in SCD are multifactorial, with no single mechanism dominating the adhesion interactions. Consequently, if a therapy is targeted to one specific interaction, vaso-occlusion may still occur through multiple other mechanisms, suggesting the need for multimodal therapy to reduce the occurrence of VOC. 20 An understanding of the pro-adhesive role of the endothelium and the inflammation resulting from ongoing hemolysis and recurrent/chronic ischemia-reperfusion injury has led to the development and investigation of newer mechanistic treatment approaches aimed at reducing their occurrence (Figure 2). New therapies are aimed at both inhibiting the adhesion of cells to the activated endothelium and preventing hemolysis and sickling through direct targeting of the hemoglobin within RBCs.
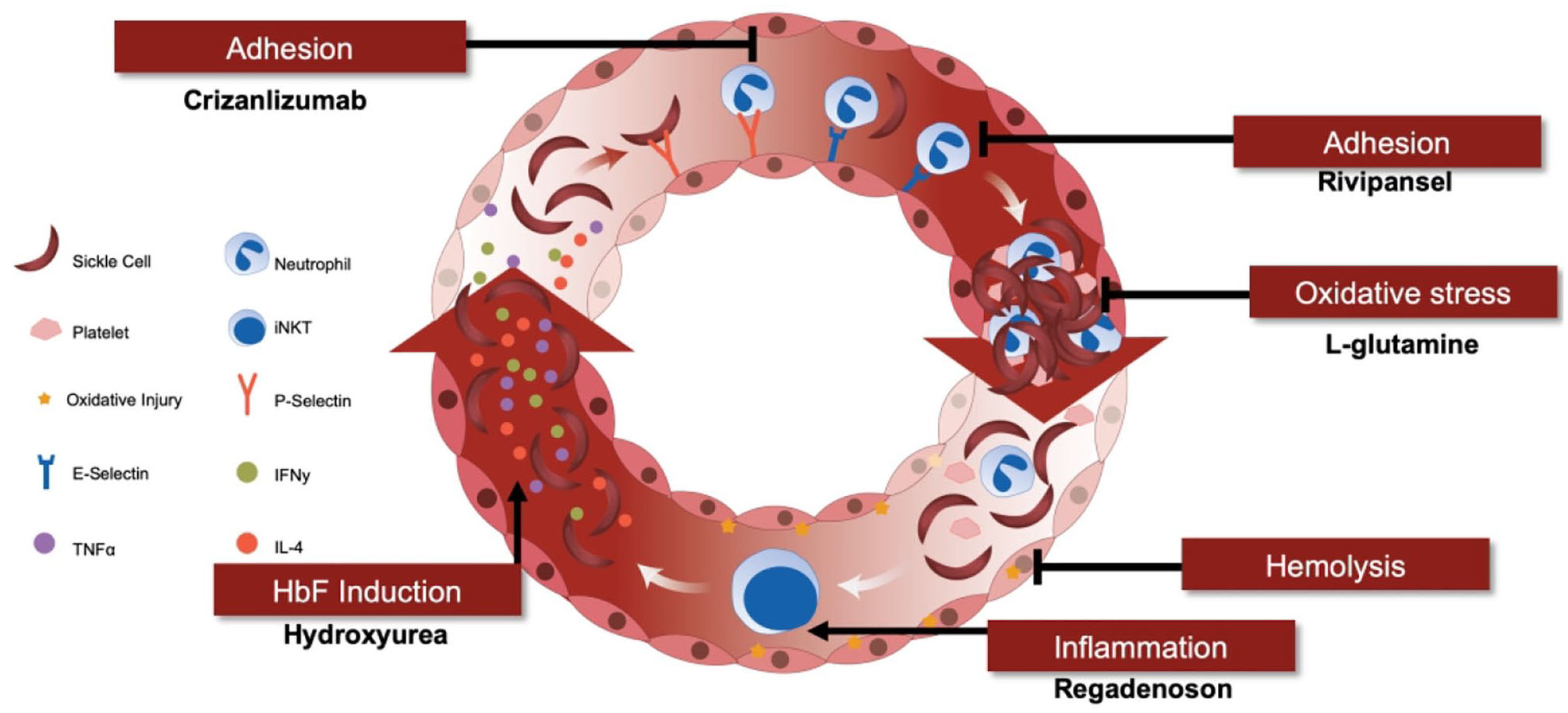
Sickle cell pain crises: role of pharmaceutical interventions in sequential pathogenic mechanisms, including adhesion, oxidative stress, inflammation, hemolysis, and HbF induction.
Current strategies to reduce the occurrence of VOCs
Prior to 2019, there were four available treatment modalities to mitigate the acute and chronic complications of SCD: hydroxyurea (HU), RBC transfusions,
Hydroxyurea
Until recently, the only medication approved to reduce the occurrence of VOCs was HU, the first drug approved by the United States Food and Drug Administration (FDA) for treatment of SCD, and transfusion therapy. HU is believed to be effective for several reasons: induction of fetal hemoglobin (HbF) to prevent RBC sickling; myelosuppression (reducing the overall number/quantity of potentially activated blood cells) leading to decreased available white blood cells, platelets, and reticulocytes; and potentially improving nitric oxide donor bioavailability. In preclinical models, HU was demonstrated to reduce the heterotypic cellular adhesive interactions, and, thus, has multiple beneficial effects in SCD.21,22
The phase III Multicenter Study of Hydroxyurea (MSH study) demonstrated significant reduction in rates of acute VOC, acute chest syndrome, and the need for transfusions among patients ⩾18 years of age with SCA (HbSS or HbSB0) receiving HU, which led to HU gaining FDA approval in 1998.23,24 In 2010, the long-term benefits and risks of HU in SCA were reported in a 17.5-year follow-up study. 25 Results showed persistently high overall mortality rates for SCD of 43.1%, but reduced mortality among those individuals with long-term HU exposure, as 87% of these deaths occurred among individuals who never took HU or had less than 5 years of exposure. The BABY-HUG trial conducted in children aged 9–18 months at randomization also showed a significant reduction in reports of pain [hazard ratio (HR) 0.59, 95% confidence interval (CI): 0.42–0.83; p = 0.002]. 26 Although HU has been available for over 20 years, there have been no high-quality studies evaluating its impact on VOC in adolescents or in patients with lifelong treatment from infancy. Furthermore, the HU studies to date have all focused on those with SCA as opposed to individuals with compound heterozygous disease (such as HbSC). Thus, the effect of HU in non-SCA SCD remains unknown.
While HU is highly effective in reducing pain crisis for some individuals with SCD, many individuals continue to have VOC despite taking HU, attesting to myriad mechanistic pathways that may be responsible for VOC that may not be fully mitigated with HU. HU also has several side effects requiring frequent monitoring, and can cause hair thinning, nail bed/skin color changes, and stomach upset. Thus, many individuals with SCD are opposed to taking HU, or are intolerant of the medication. A recent Cochrane review concluded that HU was effective in reducing pain crises for individuals with SCD based on four trials conducted in 577 individuals with SCD that compared HU and placebo. Increases in HbF and reductions in neutrophil counts were also observed. 27 Contrary to other smaller studies, this review did not show a difference between individuals receiving HU versus those receiving other treatments in terms of quality of life, deaths, or serious/life-threatening events, although the quality of evidence was low for these latter outcomes. 27 Information garnered from recent studies of other novel therapies, including SUSTAIN (crizanlizumab), DOVE (prasugrel), HESTIA (ticagrelor), and several others have demonstrated that there are still a significant proportion of patients who either are not taking HU, or experience breakthrough VOCs while taking HU.28–30
Chronic transfusion therapy
Chronic transfusion therapy is used primarily to reduce the risk of stroke in high-risk individuals with SCD.31,32 However, it can also be used to reduce the levels of circulating sickled RBCs, and thereby decrease the number of VOCs when used as prophylactic therapy. 33 Complications of chronic transfusion therapy include the risk of allo-immunization, iron overload, and its related complications. The utility of chronic transfusion therapy in SCD is limited by the availability of a compatible blood donor population. Individuals undergoing transfusion for SCD may also generate antibodies against the transfused RBCs, resulting in unpredictable, delayed hemolytic transfusion reactions and thus require phenotypically matched blood. 34
Despite improved implementation of HU and chronic transfusion therapy for disease management in SCD, recurrent VOCs continue to be associated with disease severity and mortality. Of greater concern is the increased risk of early mortality in individuals with more frequent VOCs. In a study of 264 subjects with SCD, there was a significantly younger age of death (55.8 years versus 66.2 years; p = 0.04) and a higher risk ratio (RR) of death (RR = 2.68; p = 0.03) among individuals with high rates of VOCs (defined as ⩾1 ED/hospitalization for pain in the prior 12 months) compared with those with low rates of VOCs (defined as 0 ED visits/hospitalizations in the prior 12 months). 35 In this study, 41% of participants reported HU use, and 39% reported >10 RBC transfusions during their lifetime. Major trials investigating transfusion therapy, including SIT, SWITCH, and TWITCH, showed a lower incidence of VOCs with transfusion therapy, although VOCs continued to occur despite chronic transfusion therapy.36,37
l -Glutamine
Hematopoietic stem cell transplant
HSCT is a curative approach that has traditionally been reserved only for those patients with severe SCD and complications such as stroke, recurrent acute chest syndrome, impaired neuropsychologic function, recurrent VOC, sickle lung disease, or nephropathy. 42 The first successful HSCT in a patient with SCD was reported in 1984. 43 Since that first transplant using cyclophosphamide and fractionated whole body irradiation, outcomes for HSCT have improved with the introduction of other conditioning regimens, including busulfan, fludarabine, and anti-thymocyte globulin.44,45 Despite being potentially curative, HSCT is optimized in individuals with a matched sibling donor, which is not universally available. Furthermore, the risks of HSCT, including infertility and graft-versus-host disease, have dampened enthusiasm as a universal cure. 42 As results continue to improve with this technique, including use of reduced-intensity conditioning regimens for patients with matched sibling donors and the improvement in use of alternative donors, 46 HSCT may become more widespread. A recent publication on the successes of haplo-identical transplantation has suggested this therapy may be used more in the future. 47
New approaches: recent investigations
In this review, we summarize some of the recent mechanistic approaches for the treatment of patients with SCD.
Oxygen affinity agent
Voxelotor, formerly known as GBT440, was approved by the FDA in 2019 for the treatment of SCD in adults and pediatric patients 12 years of age or older. 48 This treatment is a first-in-class orally administered agent that increases the affinity of hemoglobin for oxygen, thereby inhibiting the polymerization of HbS. 49 In a phase I/II randomized, double-blinded, placebo-controlled study, the safety, tolerability, pharmacokinetics, and pharmacodynamics of voxelotor were evaluated in subjects with SCD. Subjects treated for ⩾28 days experienced improvements in hemoglobin and mild decreases in hemolysis, and the drug was well tolerated. 49 In other studies of voxelotor in healthy volunteers (n = 40) and subjects with SCD (n = 8), the drug was well tolerated, and pharmacodynamic data established the proof-of-mechanism of voxelotor for increasing hemoglobin oxygen affinity in the RBC compartment. 50 More recent findings continue to support improvement in hemoglobin levels with voxelotor. The phase II/III HOPE study for participants aged 12–65 years was recently completed. Although the majority of participants in the study had HbSS or HbSB0 disease, the trial allowed participants with all types of SCD as long as they had a baseline hemoglobin of <10.5 /dl. 51 The study met its primary endpoint, defined as an increase in hemoglobin >1.0 g/dl at week 24, for patients taking voxelotor (51%) versus those receiving placebo (7%; p < 0.001), and this outcome was achieved regardless of concomitant HU use or severity of anemia at baseline. 51 However, the study did not meet any of the planned, required secondary endpoints, including patient-reported assessments of fatigue, pain, or reduction in number of VOCs. While there was also a trend toward a reduced incidence of VOCs in patients treated with voxelotor versus placebo, this finding was not statistically significant. There were no substantive differences in AEs related or unrelated to SCD across the treatment groups. 51 Longer-term follow-up on the impact of voxelotor on the occurrence of VOCs is awaited; a 5-year, open-label phase III extension study is currently underway [ClinicalTrials.gov identifier: NCT03573882]. As participants in this study are only those who chose to remain on therapy from the initial HOPE study, the study is biased by its design.
Fetal hemoglobin inducers
Higher levels of HbF have been shown to be protective against VOC and other pathological consequences of SCD due to its effect on inhibiting HbS polymerization.52,53 As previously discussed, HU has been shown to increase levels of HbF, demonstrating improved outcomes and lower incidence of VOCs.23,54 Other small molecule inducers have been shown to increase HbF expression for individuals with SCD. One such molecule, HQK-1001, was tested in a phase II study. Unfortunately, the results of the study were negative, with no significant increase in HbF and a trend for more pain crises in the HQK-1001 group. 55 Due to the modest impact of this agent, it is not being investigated further.
Decitabine and tetrahydrouridine
A recent phase I study examined the impact of decitabine in combination with tetrahydrouridine. 56 Results from this study showed significant increases in both HbF and total hemoglobin concentrations, and accompanying improvements in biomarkers of hemolysis, coagulation, and inflammation. The treatment was well tolerated, and the combination therapy is undergoing further clinical development for individuals with SCD [ClinicalTrials.gov identifier: NCT04055818]. To date, there are no data on the impact of decitabine plus tetrahydrouridine on clinical endpoints, including VOC.
Anti-inflammatory agents
Inflammation is a key step in the cascade of events leading to vaso-occlusion and VOC. As a result, agents targeting inflammatory processes specific to SCD as well as anti-inflammatory agents with a broader scope are of interest.
Regadenoson
Regadenoson is an adenosine A2A receptor agonist that mediates the anti-inflammatory effects of invariant natural killer T (iNKT) cells – a subset of lymphocytes that have been implicated in the pathogenesis of SCD. 57 In a phase II, randomized, placebo-controlled trial, subjects with SCD admitted to the hospital for a VOC (n = 92) were administered regadenoson at a dose known to suppress activation of iNKT cells in SCD, with the primary outcome being a >30% reduction in iNKT cells at 48 h compared with pre-dosing levels; several VOC-related secondary measures were also assessed. 57 The effect of regadenoson on the primary endpoint was not significant (p = 0.07), and regadenoson did not have an impact on the length of hospital stay, use of parenteral opioids, or visual analog assessed pain. While higher doses or earlier administration may have provided benefit in the study, iNKT cells may be more effectively depleted long-term using a monoclonal antibody-based approach to prevent VOC 58 ; alternatively, they may not play a major role in VOC pathogenesis.
NKTT120
NKTT120 is a humanized monoclonal antibody that has been shown to cause rapid and sustained deletion of iNKT cells in SCD patients without infusion-related toxicity or serious AEs in an open-label study. 58 This agent may offer the advantage of dosing every 3 months by infusion as opposed to daily dosing. Next steps for this agent are a randomized controlled clinical trial to determine its efficacy in reducing VOCs and determining the safety of prolonged iNKT cell depletion. 58
Simvastatin
The cholesterol-lowering agent simvastatin may have a beneficial impact in SCD by virtue of its effects on improving endothelial function, restoring nitric oxide production, and suppressing inflammation; although the exact mechanism of action is not clear, short-term treatment with simvastatin in patients with SCD has been shown to improve soluble biomarkers of inflammation.59,60 In a single-center pilot study of 19 individuals with sickle cell anemia, treatment with simvastatin for up to 3 months resulted in reduced sickle cell-related pain, oral analgesic use, and improvement in biomarkers of inflammation, with no simvastatin-related safety concerns or AEs reported in the study. 60 Despite the potential for myalgia associated with statin therapy, no clinical safety concerns or simvastatin-related AEs were observed. 60 Notably, the greatest reductions in pain and inflammatory markers were observed in those individuals who also received HU, suggesting a synergistic effect of combining these two therapies. 60 In light of these encouraging results, larger controlled studies are expected to evaluate the long-term efficacy and safety of simvastatin (alone or in combination with HU) in subjects with SCD.
Omega-3 fatty acids
Omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have a number of pleiotropic effects that may have a role in SCD. In addition to their anti-inflammatory effects via their role as substrates for cyclooxygenases and lipoxygenases to produce anti-inflammatory leukotrienes, thromboxane, and lipoxins, omega-3 fatty acids may also have antiplatelet and antioxidant effects, and effects on endothelial nitric oxide synthase.61,62 The membrane composition of sickled RBCs has also been shown to be abnormal, possibly contributing to their tendency to adhere to the vascular endothelium, but may be altered through omega-3 fatty acid supplementation. 63 A single-center study conducted in Sudan evaluated the impact of omega-3 fatty acid supplementation (277.8 mg DHA, 39.0 mg EPA) or placebo on VOCs in subjects with SCD aged 2–24 years (n = 128 followed up and data obtained) in a randomized, double-blind trial (n = 67 omega-3 group, n = 61 placebo group). 63 The rate of clinical VOCs (primary endpoint) was significantly reduced in the omega-3 group versus placebo (0/year versus 1/year; p < 0.0001), as were the number of days hospitalized due to VOCs (p < 0.05) and the annualized rate of VOCs (2.7/year versus 4.6/year; p < 0.001). 63
The phase II Sickle Cell Omega-3 Treatment (SCOT trial) evaluated the use of SC411, a DHA ethyl ester formulation, in a multicenter population of 67 children with SCD. 64 The trial met its primary endpoint, demonstrating a significant increase in RBC membrane DHA and EPA (p < 0.001), and the treatment was well tolerated. Sickle cell crises, analgesic use at home, opioid use, and days missed from school due to sickle cell pain were also significantly reduced with SC411 versus placebo, although the reduced rate of clinical sickle cell crises in the pooled active treatment groups versus placebo did not reach significance (rate ratio = 0.45; p = 0.07). 64 While these results need to be confirmed in a larger multicenter study, the findings suggest that omega-3 fatty acids may provide a safe, effective, and affordable option to reduce the occurrence of VOCs in individuals with SCD.
Antiplatelet agents
Platelets likely contribute to the vaso-occlusive process both as procoagulants and inflammatory mediators. The impact of modulators of platelet activation and their receptor-ligand interactions with other cells is currently under investigation to reduce vascular inflammatory processes and prevent VOCs in individuals with SCD.19,65
Prasugrel
Prasugrel is a third-generation thienopyridine that irreversibly inhibits ADP-mediated platelet activation and aggregation. This agent is approved for use in adults with acute coronary syndrome managed with percutaneous coronary intervention, and was investigated for the prevention of VOCs in the phase III, placebo-controlled DOVE trial.29,66 Children 2–17 years old with SCD (n = 341) and at least two crises in the previous year were randomly assigned to receive oral prasugrel (n = 171) or placebo (n = 170) for 9–24 months.29,66 The primary endpoint was the rate of VOCs, a composite of painful crisis or acute chest syndrome. Secondary endpoints included the rate of sickle cell-related pain and intensity of pain during VOCs, and analgesic use during VOCs, as assessed daily using pain diaries. 66 Although the rate of VOCs trended lower among those who received prasugrel (67.3%) compared with those who received placebo (72.4%), the difference was not statistically significant (events per person-year 2.30 versus 2.77). Prasugrel also had no significant impact on secondary endpoints. While the reduction in VOCs was more pronounced in those 12–17 years old and in those not receiving HU, the study was not powered to demonstrate age-specific improvement in VOCs. Nevertheless, this finding does raise the question whether antiplatelet agents might be more useful in adolescents and adults who have accumulated more vascular damage than in children with SCD. 29 A potential weakness of this study was that a lower target for platelet inhibition was chosen for this population than is typically used in adults with acute coronary syndrome; the resulting dose of drug administered may therefore have been too low to provide a significant benefit.
Ticagrelor
Ticagrelor is an inhibitor of platelet aggregation approved for use in adults with acute coronary syndrome and following myocardial infarction. 67 As with prasugrel, the rationale for the use of ticagrelor in SCD relates to the increased activation state of platelets in SCD and their further activation during VOCs. 67 In a two-part, phase II, multicenter dose-finding study (HESTIA1), a favorable dose-exposure-response relationship was demonstrated in a population of children with SCD, and the agent was well tolerated, with no increased risk of bleeding. 67 In another phase IIb study of ticagrelor in adult subjects (n = 87) with SCD (HESTIA2), the primary endpoint – mean proportion of days with pain – was reduced from baseline in all groups, including placebo, with no significant differences observed between treatments. Ticagrelor did not demonstrate any benefit on secondary pain-related and exploratory variables in the study, although the treatment was well tolerated and did not increase bleeding risk. 68 The authors suggest that larger and longer studies may be needed to determine whether platelet inhibition with ticagrelor has any beneficial impact on VOCs in subjects with SCD. A phase III study of ticagrelor (HESTIA3) evaluating its impact on reducing VOCs in pediatric patients (aged 2–18 years) with SCD versus placebo is ongoing [ClinicalTrials.gov identifier: NCT03615924]. Further studies in adults with SCD have not been planned.
Anti-adhesion agents
Molecules that inhibit selectin binding and adhesion are a potential method for reducing or improving vaso-occlusion. These molecules are especially appealing given their potential for use in combination with RBC-targeting agents described above.
Crizanlizumab
P-Selectin is one of several targets implicated in the increased adhesion activity associated with VOCs. 19 The impact of crizanlizumab, a humanized monoclonal antibody that binds P-selectin, thereby blocking its interaction with PSGL-1, has been evaluated with or without HU therapy in the phase II SUSTAIN trial. 28 In this study, subjects with SCD aged 16–65 years and 2–10 pain crises within the past 12 months (n = 198) received either crizanlizumab 5 mg/kg (n = 67), crizanlizumab 2.5 mg/kg (n = 66), or placebo (n = 65); those who were taking HU at the start of the study were allowed to continue (without dose alteration except for safety reasons), whereas those who were not taking HU could not initiate it during the 52-week study. 28 The primary endpoint was the effect on VOCs during the study period. A significant reduction in the median crisis rate/year over the treatment period was achieved with crizanlizumab 5 mg/kg (1.63) versus the placebo arm (2.98; p = 0.01), a 45.3% reduction. During the treatment phase, the crisis rate was zero in 36% of crizanlizumab 5 mg/kg, 18% of crizanlizumab 2.5 mg/kg, and 17% of placebo subjects. The secondary endpoints of days hospitalized (41.8% reduction; p = 0.45) and median time to first (4.07 months versus 1.38 months; p = 0.001) and second (10.32 months versus 5.09 months; p = 0.02) VOCs favored crizanlizumab 5 mg/kg over placebo. 28 A 62.9% reduction in the number of uncomplicated crises/year (1.08 versus 2.91; p = 0.02) was also observed for the crizanlizumab 5 mg/kg group versus placebo. Time to first VOC was also significantly increased with crizanlizumab treatment compared with placebo across all subgroups, including those with and without HU use, and those with 2–4 or 5–10 VOC events in the year prior to the study. 69 Safety findings showed a similar overall incidence of AEs and serious AEs among individuals taking crizanlizumab and placebo. An immunogenic response to crizanlizumab did not occur in the trial. A strength of the SUSTAIN trial was that it was open to patients of all genotypes and included 30% with non-HbSS genotypes. 28 Crizanlizumab was approved by the FDA in 2019 to reduce the frequency of pain crisis in adult and pediatric (aged ⩾16 years old) patients with SCD. 70
Additional agents under investigation
Vitamin D
A recent study has examined environmental and dietary factors, including vitamin D levels, and their association with crisis events in patients with SCD. The results showed that diets high in fish, milk, cheese, and eggs were associated with reduced hospital/ED visits in individuals with SCD. 71 The study also showed statistically fewer crisis-related hospitalizations for individuals with vitamin D levels >24 ng/ml; among 102 individuals with SCD, mean hospitalizations over 12 months were higher for those with severe vitamin D deficiency (0–19 ng/ml, 9 days) and mild to moderate deficiency (20–29 ng/ml, 5 days) compared with those who were not deficient (>30 ng/ml, <3 days; p < 0.05 versus severe group). 71 Another study found that high-dose vitamin D therapy in children and adolescents with SCD (n = 20) reduced the number of pain days and increased physical activity quality of life compared with patients randomized to receive placebo (n = 19). 72 Although further study is needed, the results suggest an impact of dietary factors and maintaining adequate vitamin D levels to potentially reduce the occurrence of VOCs in individuals with SCD. Despite these results, a recent Cochrane review concluded that insufficient evidence exists to support vitamin D supplementation as a means for preventing VOCs. 73
Potential targets in vascular pathways
The interaction of endothelin-1 (ET-1), a mediator of neutrophil recruitment and activation, with the endothelin B receptor (ETB) has been shown to be an important mediator of neutrophil recruitment and adhesion to activated endothelium in experimental studies of SCD mice. 74 The ET-1/ETB axis is thus under investigation as a pro-inflammatory pathway in SCD, and ET receptor inhibitors, if proven safe and efficacious in humans, may be another means of preventing VOCs in individuals with SCD. 74 Activation of the alternative complement pathway, characterized by microvascular deposition of C5b-9, has also been observed in the skin of individuals with SCD (but not healthy controls), and deposition of C3b has been observed on RBC membranes in individuals with SCD. 75 Factor H, an inhibitor of the alternative complement pathway, inhibits the interaction of sickled erythrocytes with the endothelium via interactions involving the pro-adhesive molecules Mac-1 and/or PSGL-1. The alternative complement pathway and Factor H-based inhibitors may provide another pathway of intervention to reduce the occurrence of VOCs in individuals with SCD. 75
Anti-neutrophil agents
There is evidence that a reduction in neutrophils has a beneficial impact on the occurrence of VOCs in individuals with SCD, particularly those receiving HU therapy.24,27 Thus, reducing neutrophil interactions with the activated endothelium is another avenue of investigation for the prevention of VOCs in individuals with SCD. 74
cGMP-modulating agents
Cyclic guanosine-3′5′-monophosphate (cGMP) promotes the production of HbF in erythroid cells, reduces leukocyte adhesion mechanisms, decreases endothelial adhesion molecule expression, and improves vasorelaxation. 76 cGMP production is regulated by nitric oxide binding to soluble guanylate cyclase.16,77 As already discussed, cell-free hemoglobin and arginase I reduce nitric oxide, resulting in endothelial dysfunction.13,15,16 As a result, decreased nitric oxide may result in reduced cGMP. A number of products in early clinical development have been shown to increase cGMP and therefore may have a role in mitigating the vasculopathy and nitric oxide depletion in SCD. Olinciguat stimulates soluble guanylyl cyclase, an enzyme that catalyzes the conversion of guanosine-5′-phosphate (GTP) to cGMP. 76 This compound demonstrated increased cGMP and decreased blood pressure in phase I trials with no renal clearance, suggesting it may be appropriate for use in patients with renal impairment. 76 Olinciguat is currently being studied in a phase II clinical trial in patients with SCD [ClinicalTrials.gov identifier: NCT03285178].
Another possible mechanism of cGMP in SCD is through activation of protein kinase G and induction of HbF.16,76,78 IMR-687 is a phosphodiesterase-9 inhibitor (PDE9) that acts by preventing degradation of cGMP by PDE9. It has also demonstrated induction of HbF in animal models and human cell lines. 78 Interim results from a phase II study presented at the European Hematology Association Congress in 2019 showed a trend toward increased HbF and reduced hemolysis and cellular adhesion factors in SCD patients treated with IMR-687 100 mg orally once a day. 79
cGMP-amplifying agents have been shown to intensify the nitric oxide-mediated effects of HU in animal models. 22 As a result, it is possible that all of these agents may act synergistically with HU.
Gene therapy
In several trials [ClinicalTrials.gov identifiers: NCT02140554, NCT02151526, NCT04293185, NCT02633943], gene addition therapy is performed using an ex vivo lentiviral transfer of a functional β-globin gene to hematopoietic stem cells of SCD patients is being explored as a potential therapy. 80 LentiGlobin BB305 introduces copies of an anti-sickling βAT87Q-globin gene variant into the patient’s hematopoietic stem cells. 81 Stable incorporation of the βAT87Q-globin gene variant has the potential to induce the production of functional RBCs, thereby correcting one of the main underlining causes of complications associated with SCD. 81 Proof of principle of LentiGlobin BB305 efficacy has been reported in one patient with SCD.80,81 Currently, phase I/II trials are being carried out in patients with SCD.80,81 The effect of LentiGlobin BB305 treatment on rates of VOCs and other complications associated with SCD are forthcoming.
Gene editing studies using the CRISPR/CAS-9 technology are also underway. The majority of these studies are using CRISPR to disrupt genes that fetal hemoglobin production by targeting genes such as BCL11A.82,83 These studies, also in phase I/II clinical trials, are also demonstrating very exciting potential for disease-modifying effect.
Intravenous immunoglobulin
Intravenous immunoglobulin (IVIG) inhibits Mac-1 dependent capture of RBCs by neutrophils, and has been shown to reduce neutrophil adhesion to post-capillary venular endothelium and interactions with circulating RBCs in murine models of sickle cell acute pain crisis.84–86 In a phase I study of patients with SCD with acute pain crisis, IVIG-treatment resulted in a significant decrease in Mac-1 function. 87 An ongoing randomized, placebo-controlled, phase I/II trial in SCD is ongoing to evaluate whether IVIG reduces the duration of VOC [ClinicalTrials.gov identifier: NCT01757418].
Conclusion
Acute pain remains the primary reason for which individuals with SCD currently seek acute care. While frequency, severity, and duration may vary, VOC affects many individuals with SCD.2,88 Therapies including HU,
At this time, it remains unclear if any therapy will be useful in shortening or stopping a VOC once it is initiated. Several studies have failed in this regard in recent years. While it is possible that these large studies have not met their primary endpoints due to flawed study designs and difficulty in quantifying or defining the end of a VOC, it is also possible that the vascular occlusive process cannot be altered once initiated. Indeed, the definition of pain events and pain crisis is a limitation for all of the studies of therapies discussed here, especially if dependent on patient report. This may be particularly complicated in adults who may also suffer from complex chronic pain.
As individuals are living longer with SCD, an integrative and likely multimodal approach to preventing and treating VOCs is needed. Whereas acute management of pain associated with VOCs will likely remain an essential component of treatment, the risk of opioid tolerance, reduced efficacy, and dependency over the long term remains. Furthermore, while other nonsteroidal medications are also used to decrease inflammation, their use is limited by renal dysfunction and bleeding as well as gastrointestinal complications. Other non-opioid pain therapies remain in use but have not demonstrated significant efficacy. Similarly, while they may improve patient coping/management strategies for VOCs, cognitive and/or behavioral approaches to recurrent pain associated with VOCs are limited by insufficient funding for mental health providers to administer these therapies and disparate access to sickle cell centers that would typically have these services available. Of greatest concern for the development of therapies to decrease the length of a VOC is the lack of biomarkers outside of a subjective pain report. Furthermore, there are no predictive biomarkers to identify the highest-risk subjects and/or to prioritize the use of these therapies. Lastly, although they remain to be proven in larger numbers of subjects, gene therapies have the potential to reverse or halt the disease process in SCD by introducing wild-type hemoglobin. As these therapies develop, it will be important to have initiatives that allow for greater access to specialized care and treatment once they become available.
Footnotes
Acknowledgements
Editorial support in the preparation of this manuscript was provided by Jennifer Lee of Phase Five Communications, supported by Novartis Pharmaceuticals Corporation. The authors were responsible for all content and editorial decisions and received no honoraria related to the development of this manuscript.
Conflict of interest statement
IO has received research funding from Health Resources and Services Administration, North Carolina Division of Public Health, and Patient-Centered Outcomes Research Institute; served as a consultant for Cyclerion Therapeutics Inc., Global Blood Therapeutics, Novartis Pharmaceuticals Corp., and Pfizer Inc; participated on advisory boards for Acceleron Biopharma, Forma Therapeutics, Global Blood Therapeutics, Novartis Pharmaceuticals Corp., and Pfizer Inc; participated on the speakers’ bureau for Global Blood Therapeutics, Novartis Pharmaceutics Corp., and Terumo Medical Corporation; and is a member of the Data and Safety Monitoring Board for Micelle BioPharma Inc.; currently serves as Editor in Chief of Hematology News.
DM has received research funding from bluebird bio and Global Blood Therapeutics, and served as a consultant for Global Blood Therapeutics, Novartis Pharmaceuticals Corp., and Pfizer Inc.
JK has received research funding from Health Resources and Services Administration, Center for Disease Control, and National Institute of Health as well as from bluebird bio and Novartis Pharmaceuticals Corp.; served as a consultant for bluebird bio, Imara Inc., Modus Therapeutics, Novartis Pharmaceuticals Corp., Agios, Beam Therapeutics, and Sanofi; and received honoraria from Bluebird Bio, Global Blood Therapeutics and Terumo Medical Corporation.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.