
Abstract
Myelodysplastic syndromes (MDS) are clonal hematopoietic stem cell disorders characterized by ineffective hematopoiesis with peripheral blood cytopenias, dysplastic cell morphology, and a variable risk of progression to acute myeloid leukemia (AML). The hypomethylating agents (HMA) azacitidine and decitabine have been used for over a decade in MDS treatment and lead to a modest survival benefit. However, response rates are only around 40% and responses are mostly transient. For HMA-refractory patients the prognosis is poor and there are no therapies approved by the United States Food and Drug Administration.
Combinations of HMAs, especially along with immune checkpoint inhibitors, have shown promising signals in both the frontline and HMA-refractory setting. Several other novel agents including orally available and longer acting HMAs, the BCL-2 inhibitor venetoclax, oral agents targeting driver mutations (IDH1/2, FLT3), immunotherapies, and new options for intensive chemotherapy have been studied with variable success and will be reviewed herein. Except for the minority of patients with targetable driver mutations, HMAs – likely as part of combination therapies – will remain the backbone of frontline MDS treatment. However, the wider use of genetic testing may enable a more targeted and individualized therapy of MDS patients.
Introduction
Myelodysplastic syndromes (MDS) constitute a heterogenous group of clonal hematopoietic stem cell disorders characterized by ineffective hematopoiesis leading to peripheral blood cytopenias, dysplastic cell morphology, and an increased risk of progression to acute myeloid leukemia (AML).1–3 Individual disease courses are highly variable, and treatment approaches need to be tailored to a patient’s symptom burden, risk of progression to AML, and comorbidities. 4 The most commonly used risk stratification tools for MDS patients are the International Prognostic Scoring System (IPSS), its revised version (IPSS-R), and the World Health Organization (WHO)-based prognostic scoring system (WPSS), which all classify patients into categories from very low to very high risk based on (1) cytogenetic features (karyotype, presence of recurrent cytogenetic abnormalities), (2) bone marrow blast percentage, and (3) the extent/degree of peripheral blood cytopenias.5–8
For patients with lower-risk MDS, clinical treatment focuses on management of symptoms resulting from peripheral blood cytopenias and minimizing the need for transfusions, with a wide array of treatment options available ranging from erythropoiesis-stimulating agents to immunosuppression, lenalidomide, hypomethylating agents (HMA), and TGF-β pathway inhibitors.4,8–10 While the median overall survival (OS) for lower-risk MDS (IPSS-R score < 3) has been reported to be 5.3 years (without therapy), OS for patients with intermediate- or higher risk MDS is poor, with only 0.8 years (without therapy) in the very-high risk strata. 5 Given this dismal prognosis, more aggressive upfront approaches are warranted, including allogeneic hematopoietic stem cell transplant (allo-SCT) for medically fit high risk MDS patients.
The HMAs 5-azacytidine (AZA) and its analogue 5-aza-2′deoxycytidine (decitabine; DEC) are both approved by the United States (US) Food and Drug Administration (FDA) for the treatment of MDS. AZA has also been approved in Europe based on the results of the MDS AZA-001 trial. 11 The overall response rate (ORR) ranges between 25% and 40%, and there is a significant reduction of the risk of progression to AML. A 7-day regimen of IV/SC AZA at 75 mg/m2 every 28 days demonstrated a 9.5 month OS benefit in patients with higher-risk MDS based on IPSS in comparison with conventional care options.11–15 Subsequent real-world data have nuanced these results with lower benefits of HMA compared with the original studies, which might be due to differences in adherence to dosing schedules, treatment duration, and less rigorous patient selection compared with the landmark clinical trials.16,17
For patients progressing on HMA therapy, the prognosis is poor, with a median OS of 4–5.6 months for higher risk MDS patients (and 14–17 months for low risk MDS patients).18–20 It is important to note that the definition of HMA failure occurs along a spectrum reaching from the mere absence of hematologic improvement to progression to higher-risk MDS and/or AML. While patients with lower-risk MDS can remain within the lower-risk MDS category with an array of additional treatments available, patients progressing to higher-risk MDS or AML have limited options especially if they are not eligible for intensive chemotherapy or allo-SCT. This underscores the need for additional therapeutic options. 18 In this review, we outline the current role of HMAs in MDS treatment, highlight the rationale and available clinical data for combination therapy with HMA and various other agents, and outline the research agenda and potential novel treatment options.
Hypomethylating agents in MDS treatment
Hypomethylating agents as epigenetic modifiers in myeloid neoplasms
Gene expression is highly dynamic and tightly regulated by various epigenetic processes that modify the interaction between the DNA molecule and its histone protein scaffold.21,22 Two of the major epigenetic modification processes are DNA methylation and hydroxymethylation. Mutations affecting genes involved in both DNA methylation (e.g., DNMT3A) and demethylation (e.g., TET2) have also been found in >10% of patients with MDS and AML.23,24 Methylation of CpG islands in the DNA leads to the suppression of gene transcription by preventing the binding of transcription factors to the DNA strand. 22 Mutations affecting DNA methylation can cause malignant transformation by the silencing of tumor suppressor genes, and have been shown to be an early step in the genetic evolution of myeloid neoplasms.25,26 The presence of either one or both epigenetic silencing and/or genetic mutations confers a predisposition to MDS and its clinical phenotype.
The HMAs AZA and DEC [as well as guadecitabine (SGI-110; a decitabine analogue that is not metabolized by cytidine deaminase and therefore has an extended half-life)] act as DNA methyltransferase inhibitors and lead to demethylation of DNA in a cell-cycle-dependent manner. 27 This has been shown to restore the transcription of previously silenced genes and leads to clinical benefit in patients with myeloid neoplasms.11,15,28,29 Selected clinical trials of HMA monotherapy in MDS are summarized in Table 1.
Selected clinical trials of HMA monotherapy.

AML, acute myeloid leukemia; AZA, azacytidine; CMML, chronic myelomonocytic leukemia; CR, complete remission; DEC, decitabine; HI, hematologic improvement; HMA, hypomethylating agents; MDS, myelodysplastic syndrome; mCR, marrow CR; ORR, overall response rate; OS, overall survival; Ref, reference; RR, relapsed/refractory.
Current recommendations for HMA therapy in MDS
While HMA therapy is approved only for IPSS higher-risk MDS patients in Europe, AZA and DEC are approved for the treatment of all patients with MDS in the US. 17 The National Comprehensive Cancer Network (NCCN) is recommending HMA use primarily for patients with intermediate- or high-risk MDS who are not candidates for intensive therapy, who are unlikely to respond to other treatment modalities (e.g., immunosuppressive therapy), or as a bridge to allo-SCT. 8
Given their cell-cycle-dependent mechanism of action, these agents must be administered on a daily basis for at least 5 days and 7 days at 4-week intervals for at least 4–6 cycles, before response can be assessed. 8 The 7-day continuous administration schedule of AZA can be cumbersome due to logistical challenges of a therapy that includes a weekend (or 2 days of the following week). However, in a randomized trial by Lyons et al., the response rates were similar between the 7-day, 5-2-2 (five consecutive doses followed by 2 days off and two additional doses) and 5-day schedules. 34 A similar trial to identify the optimal administration schedule was conducted for DEC and showed that the 5-day intravenous administration schedule of 20 mg/m2 was superior to both 5-day subcutaneous administration of 20 mg/m2 and 10-day intravenous treatment with 10 mg/m2 in terms of complete remission rate (39% versus 21% versus 24%, respectively; p < 0.05). 30
Most patients who eventually respond to HMAs will do so within the first six cycles (and even four cycles can lead to a survival advantage). Thus, it is recommended to complete at least six cycles of AZA before declaring that a MDS patient is HMA-refractory.8,35 However, for patients responding to HMAs, this treatment should be continued until disease progression as studies have shown that responses may continue to improve with continued therapy, 36 and that treatment interruptions even in patients with CR can lead to rapid relapses which are often resistant resumption of HMA therapy. 37
Treatment options for the HMA resistant patient
Since the prognosis of patients failing HMA therapy is dismal except for the small minority of patients eligible for allo-SCT, there is an urgent need for both prevention of HMA failure by (1) optimization of frontline therapies (e.g., adding synergistic agents to HMA therapy) and/or (2) improved salvage therapies for HMA refractory MDS patients.
Despite the frequency and high clinical relevance of HMA resistance, no formal recommendations from the NCCN or the European Leukemia Net for this scenario exist. This is further complicated by the fact that HMA failure is not a homogenous definition but occurs along a spectrum reaching from failure to achieve hematologic improvement to progression to higher risk MDS and AML. Subsequent treatment selection should be individualized and guided by the patient’s IPSS-R risk category (lower-risk versus higher-risk), comorbidities and patient preference/goals. Various expert recommendations have been published recently.17,38 Furthermore, novel agents in MDS therapy have often been extrapolated from active agents for the treatment of AML. While both disorders are related, the direct applicability of AML study results to MDS patient cohorts cannot be assumed. Rather, this emphasizes the need for dedicated clinical trials specific to the MDS setting in order to generate high quality clinical evidence and to inform treatment decisions specific to the MDS phenotype.
Both frontline and salvage therapy of MDS patients require appropriate assessment of their individual risk (IPSS or IPSS-R) and their personal treatment goals (option for curative intent possible or not possible versus treatment of symptoms versus treatment to delay time to progression, etc.). However, standard risk stratification tools such as IPSS and IPSS-R have limited predictive value in the HMA-refractory setting. Therefore, a dedicated risk stratification tool for patients with HMA-refractory MDS has recently been proposed and validated. It includes patient (age, performance status) and disease characteristics (complex cytogenetics (>4 abnormalities), bone marrow blast percentage >20%, platelet count, and red cell transfusion dependency) to stratify patients into low-risk and high-risk categories with median OS of 11.0 months and 4.5 months, respectively.39–41
It has been shown that up to 77% of lower-risk MDS patients progressing on HMAs remain in the lower-risk group, with various treatment options being available based on cytogenetics, comorbidities, and patient preference. 18 This may be due to the mutational spectrum of these cohorts with spliceosomal mutations being predominant.
Lenalidomide
In the subpopulation of MDS patients with del(5q), lenalidomide is a highly effective treatment in the first-line setting to decrease transfusion dependence but data from patients with del(5q) HMA-refractory disease show that lenalidomide salvage therapy is only of limited clinical benefit. In a small retrospective study of 10 patients, 60% (three out of five) of patients with del(5q) who progressed on HMAs responded to subsequent treatment with lenalidomide. 42 In a phase II trial of 24 unselected HMA-refractory patients, marrow CRs (mCR) were seen in 33% and hematological improvement (HI) in 8% of patients, respectively, with a median OS of 106 days. 43 However, in patients with non-del(5q) MDS lenalidomide yielded response rates of only around 10% and was associated with significant adverse events.44,45 Therefore, lenalidomide should be given rather as frontline treatment in patients with transfusion dependent MDS and del(5q) and does not have a role in HMA-refractory cases.
Intensive chemotherapy
For medically fit patients, intensive treatment with induction chemotherapy can be considered and is frequently used as a bridge to allo-SCT. In a recent international multicenter retrospective analysis of 307 MDS patients failing HMA, three intensive induction chemotherapy regimens were compared (7+3, intermediate- to high-dose cytarabine, and purine nucleoside analogue-based regimens). The ORR was 41% with a median OS of 10.8 months with 40% of patients proceeding to allo-SCT. 46 Of note, there was no statistically significant difference in terms of median OS between the three tested chemotherapy regimens. 46
MDS patients with progression to AML are eligible for treatment with CPX-351, a 5:1 liposomal formulation of cytarabine and daunorubicin. CPX-351 was tested in an open-label, randomized, phase III trial of 309 patients with newly diagnosed high-risk or secondary AML aged 60–75 years. 47 CPX-351 significantly improved median OS and yielded higher overall remission rates compared with standard 7+3 induction chemotherapy [median OS: 9.56 versus 5.95 months (one-sided p = 0.003); ORR: 47.7% versus 33.3% (two-sided p = 0.016)]. 47 While CPX-351 is FDA-approved only for the treatment of newly diagnosed therapy-related AML or AML with myelodysplasia-related changes, about 50% and 30% of patients in the phase III trial by Lancet et al. had preceding MDS and were treated with HMA before, respectively. 47 A recent multicenter analysis of patients with secondary AML who had received HMA prior to AML transformation showed that CPX-351 led to similar rates of complete remission (CR) and CR with incomplete count recovery (CRi) compared with 7+3 (41.1% versus 32%; p = 0.526). Of note, patients who had received more than four cycles of HMA prior to AML transformation were significantly less likely to respond to CPX-351 compared with patients with no HMA exposure (25.0% versus 64.3%; p = 0.04). 48 Several ongoing clinical trials investigating CPX-351 (modified lower doses for MDS) in the HMA-refractory high-risk MDS patient population are currently ongoing [ClinicalTrials.gov identifiers: NCT03957876; NCT03896269].
However, emerging data suggest that treatment decisions regarding intensive chemotherapy should not only be based on a patient’s “fitness” for chemotherapy but could be supplemented by molecular and cytogenetic features. For example, intensive chemotherapy and clofarabine in combination with low-dose cytarabine might be an effective alternative in patients with NPM1 mutations or a diploid karyotype, respectively.49,50 Conversely, a complex karyotype and the presence of TP53 mutations have been associated with lower-response rates to intensive chemotherapy among MDS and AML patients.51–53 However, additional studies are needed to validate treatment selection based on molecular testing.
Allogeneic hematopoietic stem cell transplant
Allo-SCT remains the only potentially curative treatment modality for MDS, and should be considered for eligible patients with higher-risk by IPSS-R or in lower-risk patients with adverse prognostic factors such as high transfusion burden, profound cytopenias, or poor cytogenetic features. 13 Among 6434 patients with MDS or secondary AML enrolled in the registry of the European Society for Blood and Bone Marrow Transplantation, 5-year and 10-year OS rates were 43% and 35%, respectively, with non-relapse-related mortality (NRM) in 34% of patients after 10 years. 54 Although age was associated with an excess mortality, this was partly attributable to the age-related population mortality and rates of allo-SCT in MDS patients ⩾65 years of age continue to increase. 54
A careful selection of patients based on patient (age, comorbidities) and disease characteristics (IPSS-R score, bone marrow blast percentage, cytogenetic and molecular features) is necessary to achieve the optimal balance between risks and benefits and various prognostic tools have been developed.13,55,56 Emerging data suggest that age should not be used as the only factor to determine transplant eligibility with studies supporting the safety and efficacy of reduced-intensity conditioning regimens and comparable rates of OS and NRM in patients ⩾65 years and younger patients.57,58
The optimal timing of allo-SCT is controversial but it should be considered in patients with relapsed or refractory disease following frontline intensive chemotherapy or HMA. 13 Among 37 patients proceeding to allo-SCT after HMA-failure in the study by Prebet et al., median OS was 19.5 months, supporting its role in selected patients with HMA failure. 19 However, it is important to note that pre-transplant blast burden has a significant impact on outcomes following allo-SCT and cytoreductive treatment with either intensive chemotherapy or HMA to achieve ideally less than 10% blasts is recommended without clear evidence favoring either intensive chemotherapy or HMA.13,59,60 While additional studies to determine the optimal timing, patient selection, donor source, and conditioning regimen are needed, all patients with higher-risk MDS and selected lower-risk MDS patients should be evaluated for allo-SCT, especially after failure of frontline treatment.
For patients who are not eligible for intensive treatment several, still largely experimental treatment options that entail novel HMAs, combination therapy with venetoclax or immunotherapy, and targeted therapies for patients with certain driver mutations are also being actively investigated. Table 2 provides an overview of ongoing trials with investigational agents in MDS patients with HMA failure.
Selected ongoing trials of experimental agents in HMA-failure MDS.
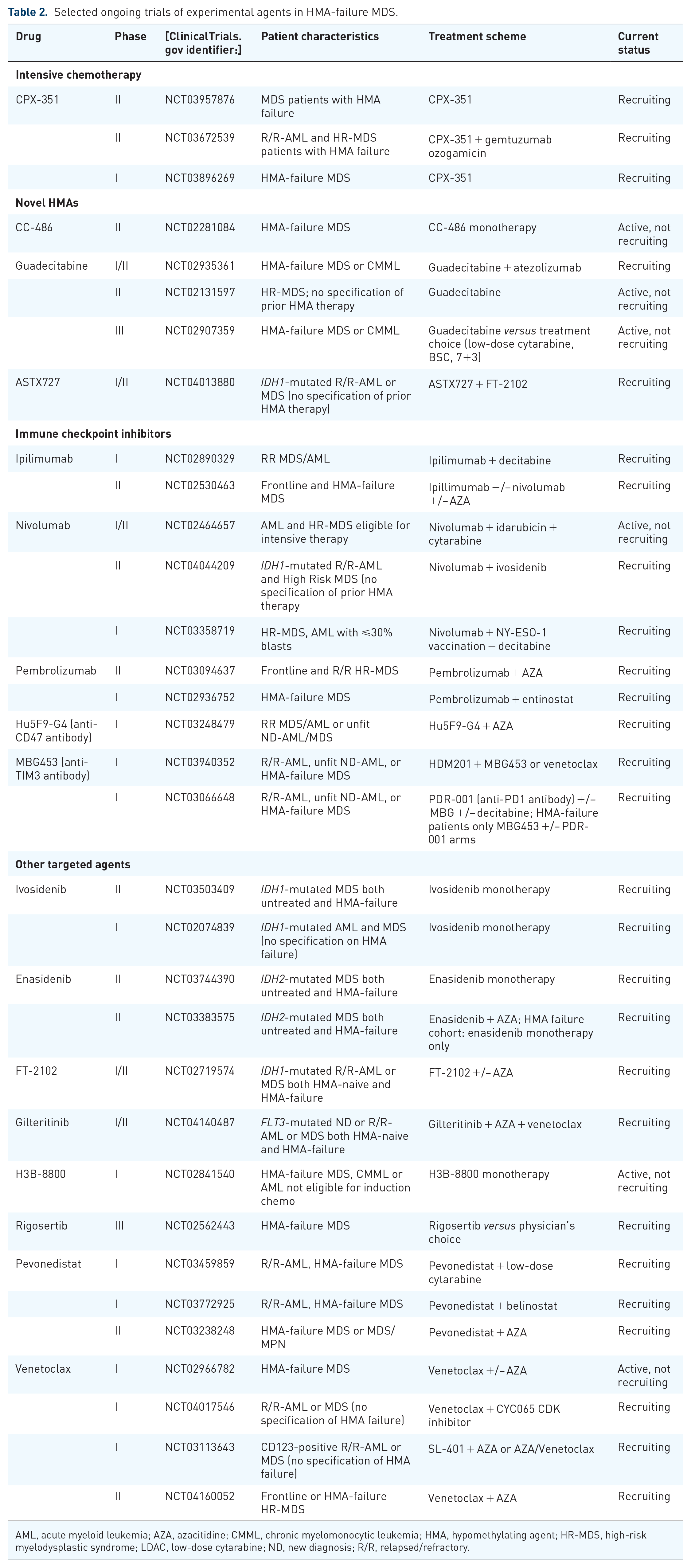
AML, acute myeloid leukemia; AZA, azacitidine; CMML, chronic myelomonocytic leukemia; HMA, hypomethylating agent; HR-MDS, high-risk myelodysplastic syndrome; LDAC, low-dose cytarabine; ND, new diagnosis; R/R, relapsed/refractory.
Novel HMAs
As mentioned previously, AZA and DEC have a fairly short half-life, which may limit their biologic activity. This has led to the development of guadecitabine (SGI-110) – a DEC analogue resistant to deamination by cytidine deaminase, and maybe therefore more effective and easier to administer given the less frequent dosing requirements than AZA and DEC. 61 Single arm phase I/II studies of guadecitabine in both first-line and relapsed/refractory (R/R) AML and MDS have shown ORRs of 8.6% (2 CR, 3 CRi, 1 PR in 74 AML patients, 2 CRs in 19 MDS patients) in the pretreated setting and 30–50% in the frontline setting, respectively.29,31,61,62 Guadecitabine appeared to be well-tolerated, with the most common grade 3 or greater non-hematologic adverse effects in these trials being febrile neutropenia (31–66%), pneumonia (27–36%), and sepsis (16–27%). Several dosing schedules were tested with a 5-day subcutaneous administration regimen of 60 mg/m2 emerging as the most effective and best tolerated dose.29,31,61,62 Of note, in a phase I/II study [ClinicalTrials.gov identifier: NCT01261312] testing guadecitabine in 105 MDS patients (51 treatment-naïve and 54 HMA-refractory patients) guadecitabine yielded ORRs of 40% and 55% in the frontline and 51% and 43% in the HMA-refractory setting when used at 60 mg/m2 and 90 mg/m2, respectively. 31 Two deaths (one each due to septic shock and pneumonia) in this study were deemed to be treatment related. 31 The notably high response rates in the HMA-refractory setting suggest that guadecitabine might be a viable option in this context. This led to a phase III clinical trial that is randomizing patients with MDS or chronic myelomonocytic leukemia (CMML) to either 60 mg/m2 guadecitabine or treatment choice [low-dose cytarabine (LDAC), standard induction chemotherapy, or best supportive care]. The trial is currently recruiting patients and no results have been published yet [ClinicalTrials.gov identifier: NCT02907359; ASTRAL-3].
However, keeping the limitations of extrapolating results from AML studies to MDS patients in mind, a recently presented randomized phase III trial [ClinicalTrials.gov identifier: NCT02348489; ASTRAL-1] comparing guadecitabine to treatment choice (AZA, DEC, LDAC) in elderly, treatment-naïve AML patients failed to meet its primary endpoint of improved survival and CR rate with guadecitabine (median OS 7.1 months versus 8.5 months; p = 0.73; CR rate 19.4% versus 17.4%, p = 0.48, for guadecitabine and physician choice, respectively). 63
ASTX727 has been developed as an oral combination drug of cedazuridine – a cytidine deaminase inhibitor– with decitabine. Preliminary data from a phase II study of 50 patients with intermediate- or high-risk MDS or CMML of whom 94% had not been previously exposed to HMAs showed an ORR of 62% with 8 (16%) CR, 14 (28%) mCR, and 9 (18%) HI. 32 The most common adverse events of grade 3 or greater were hematologic (neutropenia 48%, thrombocytopenia 38%, anemia 22%, leukopenia 20%), febrile neutropenia 38%, and pneumonia 20%. 32 A phase III, open-label crossover study of ASTX727 versus IV decitabine in MDS and CMML patients is currently ongoing [ClinicalTrials.gov identifier: NCT03306264; ASCERTAIN trial]. Preliminary data from 101 evalauble MDS and CMML patients showed ORR (CR + PR + mCR + HI) of 64.4%, with a CR rate of 12% and pharmacokinetic and pharmacodynamic data showing equivalence of ASTX727 and IV decitabine as determined by the extent of DNA demethylation. 64
In order to increase patient’s quality of life by reducing the burden of frequent office visits for HMA injections and injection site reactions, as well as prolonging drug exposure time, an oral formulation of AZA, known as CC-486, has been developed and tested in various trials.33,65,66 These trials established the safety and potentially increased therapeutic efficacy of an extended dosing schedule (either 14 days or 21 days of 300 mg CC-486 daily per 28-day cycle) with response rates of up to 46% and an acceptable toxicity profile (grade 3–4 adverse events in up to 83%) with gastrointestinal (GI) toxicity being the most common non-hematologic adverse event, and rates of febrile neutropenia of up to 42%.33,65,66 However, additional studies are needed to further define the role of CC-486 in the treatment landscape of MDS, with one clinical trial of CC-486 in HMA-refractory MDS patients being currently active [ClinicalTrials.gov identifier: NCT02281084]. Maintenance therapy with CC-486 in AML patients in first remission following induction chemotherapy has been studied in the randomized phase III QUAZAR AML-001 trial. 67 Emerging data from this trial using a 14-day 300 mg dose of CC-486 as maintenance therapy in de novo AML patients in first CR showed a 9.9 month OS benefit with CC-486 compared with placebo [24.7 months versus 14.8 months; HR: 0.69; (95% CI: 0.55–0.86); p = 0.0009] and might foretell a promising outlook for its use as post-induction chemotherapy maintenance for MDS patients. 67
Combination therapy
While the activity of HMAs as single agents is limited, several combination therapies utilizing the synergistic effects of HMA when combined with intensive chemotherapy, other forms of epigenetic therapy [histone deacetylase (HDAC) inhibitors], the BCL-2 inhibitor venetoclax and immune checkpoint inhibitors (ICI) have been tested and yielded impressive results. 26 Table 3 summarizes selected ongoing and completed clinical trials of combination therapies of HMAs in myeloid neoplasms.
Selected clinical trials of HMA in combination therapy.

AML, acute myeloid leukemia; AZA, azacitidine; CMML, chronic myelomonocytic leukemia; CR, complete remission; Cri, complete remission with incomplete cell count recovery; DEC, decitabine; HI, hematologic improvement; HMA, hypomethylating agent; HR-MDS, high-risk myelodysplastic syndrome; LDAC, low-dose cytarabine; ND, new diagnosis; ORR, overall response rate; OS, overall survival; Ref, reference; R/R, relapsed/refractory; s-AML, secondary AML; SD, stable disease.
Combination of HMAs with venetoclax
B-cell leukemia/lymphoma-2 (BCL-2) is an anti-apoptotic protein that is overexpressed in various hematologic malignancies including AML and MDS, and inhibits cell death by blocking permeability of the mitochondrial outer membrane.79,80 It has also been repeatedly implicated in leukemic stem cell survival – a cell population that persists after chemotherapy and gives rise to disease relapse.80,81
Preclinical studies have shown that BCL-2 inhibitors can act as sensitizing agents making leukemic cells more susceptible to HMAs. 82 In addition, treatment with HMAs has been shown to increase the levels of BCL-2, which has been linked to resistance to HMAs and chemotherapy. 83 Therefore, combining the oral BCL-2 inhibitor venetoclax with HMAs uses synergistic mechanisms of action to target resistance mechanisms that have limited monotherapy with either of these agents. 68 Notably, a recent in vitro study has also shown that the combination of venetoclax and AZA is effective even at lower doses of AZA, which potentially allows specific targeting of leukemic cells while sparing normal hematopoiesis. 84
Venetoclax has recently been approved by the FDA for the treatment of newly diagnosed AML in combination with AZA, DEC, or LDAC for patients who are age 75 years or older, or who are ineligible for intensive chemotherapy based on a phase I/II clinical trial of venetoclax + AZA or DEC that showed a CR + CRi rate of 73% with a median OS of 17.5 months. 85 Similar but slightly inferior results have also been published for the combination of LDAC with venetoclax in 82 AML patients older than 60 years of age of whom 49% had secondary AML and 29% had prior HMA treatment [54% CR/CRi; median OS 10.1 months (95% CI, 5.7–14.2)]. 86 Preliminary data from a phase Ib study [ClinicalTrials.gov identifier: NCT02942290] in previously untreated MDS patients have recently been presented. 70 Among 57 evaluable patients treated with AZA + venetoclax, 18 (31.6%) and 22 (38.6%) patients achieved a CR and mCR, respectively, with an 18-month OS estimate of 74% (95% CI: 50–87%). 70 However, adverse events were common with neutropenia (61%), thrombocytopenia (39%), leukopenia (31%), and anemia (20%) being the most common grade 3/4 adverse events. 70 Febrile neutropenia occurred in 31% of patients with four deaths adjudicated to infection complications. 70
In patients with HMA-refractory MDS, data on the efficacy of the combination of venetoclax with AZA, DEC, or LDAC are scarce. Two retrospective analyses of salvage regimens of venetoclax and low-intensity chemotherapy in myeloid neoplasms showed ORR of 22% and 28.6%, respectively.87,88 However, these studies included only two and one MDS patient, respectively. Therefore, dedicated studies in HMA-refractory MDS patients are needed, and larger prospective studies combining venetoclax with DEC [ClinicalTrials.gov identifier: NCT03404193] and AZA [ClinicalTrials.gov identifier: NCT02966782] in R/R myeloid malignancies are currently ongoing. Preliminary results from a phase II trial of patients with HMA-refractory MDS treated with venetoclax monotherapy or venetoclax + AZA have been presented recently. 71 While venetoclax monotherapy had only very modest efficacy [16 out of 22 patients evaluable, 1 mCR, median PFS: 3.4 months (95% CI: 1.9–5.2 months), 57% 6-month OS estimate (95% CI: 22–81%)], the ORR in the combination arm was a promising 50% [12 out of 24 patients, 3 patients (12%) with CR, 9 patients (38%) with mCRl; 6-month PFS: 76% (95% CI: 50–89%), 9-month OS: 83% (95% CI: 55–95%)]. 71 Of note, based on the studies in AML patients as well as early data from MDS patients, the combination of AZA and venetoclax leads to significant myelosuppression, and careful patient selection and monitoring (and dose adjustments) are warranted.
Combination of HMA with ICIs
Epigenetic silencing of genes regulating effector T-cell function has been shown to contribute to the immune system evasion of leukemic cells and the immunosuppressive tumor microenvironment, which are essential for tumor cell survival. 89 The use of ICI targeting programmed cell death (PD)-1, its ligand (PD-L1), and cytotoxic T-lymphocyte associated protein (CLTA)-4 as monotherapy in myeloid neoplasms is limited by the low expression of these ICI molecules on leukemic blasts and immune evasion due to the upregulation of additional inhibitory checkpoint receptors.90,91 However, the combination of HMAs with ICI may have synergistic effects due to the increased expression of leukemia-associated antigens such as NY-ESO-1 and MAGE-A, MHC-I and other co-stimulatory molecules (ICAM, CD80, CD86), which can stimulate a more potent anti-leukemia immune response.92–97 Furthermore, resistance to HMA monotherapy can be explained by the increased expression of PD-1/PD-L1 and CTLA-4 during treatment with HMA.98–100 Therefore, the combination of ICI with HMA may overcome these resistance mechanisms and provide a synergistic effect.
Encouraged by preclinical experiments that supported the presumed synergy between HMAs and ICI, 101 various clinical trials that combine HMAs with various PD-1 inhibitors, PD-L1 inhibitors, or the CTLA-4 inhibitor ipilimumab have been conducted or are currently ongoing (Tables 2 and 3). While clinical data available to date are rare, preliminary data of a phase II study of nivolumab, an anti-PD1 antibody, or ipilimumab in combination with AZA in MDS patients available in abstract form are encouraging. 73 In 76 patients with MDS (54% front-line and 46% HMA-refractory) treated with either AZA + nivolumab, AZA + ipilimumab, nivolumab alone, or ipilimumab alone, ORR and median OS showed synergistic effects of AZA + nivolumab or ipilimumab (ORR in 15/20 (75%), 15/21 (71%), 2/15 (13%), and 7/20 (35%) of patients with median OS of 12 months, not reached, 8 months, and 8 months treated with AZA + nivolumab, AZA + ipilimumab, nivolumab alone, or ipilimumab alone, respectively). 73 Importantly, the safety profile for the combination therapy appeared manageable, although data on this is limited and further research is needed to define the role of this potentially promising therapeutic option in the MDS treatment landscape.
Another clinical trial in 70 RR-AML patients treated with AZA and nivolumab demonstrated a 33% ORR (58% in frontline and 22% in HMA-pretreated patients) and a median OS of 6.3 months, which appears superior to historic controls of AZA monotherapy. 102 However, 11% of patients developed grade 3/4 immune-related adverse events (irAE), which were controlled with corticosteroids except for two cases of irAE-related deaths from pneumonitis and hemophagocytic lymphohistiocytosis that were refractory to steroids and infliximab. 102
However, more recent publications for frontline treatment of AML and higher-risk MDS have had only lackluster results. In a randomized phase II trial of AZA in combination with the anti-PD-L1 antibody durvalumab in AML patients ineligible for intensive chemotherapy (n = 129 patients) and patients with higher-risk MDS (n = 84 patients), the combination arm did not improve ORR and median OS compared with AZA monotherapy in both AML and MDS patients. 103 A smaller single arm study of AZA in combination with pembrolizumab on the other hand, did show moderate clinical activity of this combination in R/R-AML patients (57% with prior HMA therapy) with an ORR of 24% and CR/CRi rate of 11%. 104 However, how this translates to MDS patients and whether these results can be verified in larger, controlled trials needs to be further evaluated.
Research on the combination of AZA and ICI is ongoing, and current clinical trials also investigate the combination of nivolumab and ipilimumab with AZA [ClinicalTrials.gov identifier: NCT02397720]. Besides PD-1 and CTLA-4, there are several other co-inhibitory T-cell receptors that can be therapeutically targeted, such as T-cell immunoglobulin mucin (TIM)-3 and lymphocyte activation gene (LAG)-3. 105 Given that the coexpression of TIM-3 and PD-1 by T-cells in bone marrow aspirates from AML and MDS patients has been associated with immune exhaustion and higher rates of relapse after allo-SCT, combined inhibition of TIM-3 and PD-1 might be a powerful therapeutic concept that is currently tested in a phase I study [ClinicalTrials.gov identifier: NCT03066648].90,106 Hoping to enhance the therapeutic benefit by synergistic effects of HMA and ICI, another arm of this study is combining the anti-TIM-3 antibody MBG453 with DEC. Preliminary data from the combination arm (DEC + MBG453) of this study showed a rate of CR/CRi in 50% of higher-risk MDS patients (n = 16 patients) with eight patients (14%) developing ⩾grade 2 suspected irAEs considered to be treatment-related. 107 However, the efficacy of this combination in the HMA-refractory setting needs to be further elucidated.
Promising data for the frontline treatment of MDS have also been published for the combination of AZA with the anti-CD47 antibody magrolimab [ClinicalTrials.gov identifier: NCT03248479]. 108 CD47 is an inhibitory immune checkpoint on macrophages inhibiting phagocytosis and has been shown to be upregulated by leukemic stem cells enabling immune escape and whose upregulation has been associated with adverse outcomes in AML patients.109,110 Inhibiting CD47-mediated immune escape could therefore lead to enhanced phagocytosis of leukemic stem cells and AML blasts. Preliminary data from a phase Ib trial of 68 HMA-naive patients (39 MDS and 29 AML) treated with magrolimab + AZA showed an ORR of 91% (30 out of 33 evaluable patients) in MDS with 42% CR rate, a median duration of response that has not been reached at 5.8 months of median follow up, and a 100% 6-month OS estimate. 108 Notably, even in patients with TP53 mutations, ORR of 75% in both the AML and MDS patient cohort with 6-month OS estimates of 91% and 100%, respectively, have been shown. 108 Since CD47 is also expressed on erythrocytes and erythrocyte precursor cells, hemolytic anemia can be an on-target, off-leukemia adverse event seen with magrolimab. 111 However, the combination of AZA + magrolimab appeared to have a safety profile similar to that of AZA monotherapy with anemia (38%), fatigue (21%), neutropenia (19%), and thrombocytopenia (18%) being the most common treatment-related adverse events with only one patient discontinuing treatment due to adverse events. 108 Those promising results have led to a randomized phase III trial comparing AZA + magrolimab with AZA + placebo in untreated MDS patients [ClinicalTrials.gov identifier: NCT04313881].
Other combination therapies
In addition to ICI and venetoclax, conventional chemotherapy, targeted agents (e.g., FLT3- and IDH1/2 inhibitors), and HDAC inhibitors have been used as combination therapies with HMAs. However, most of these studies have been conducted in AML patients and have been recently reviewed elsewhere in greater detail. 26 However, none of these combinations has yielded as impressive results as the combinations of HMAs with venetoclax and ICI thus far. For example, the addition of AZA to cytarabine/daunorubicin (“7+3”) induction chemotherapy could potentially increase the susceptibility of leukemic cells to chemotherapy by increasing the expression of tumor suppressor genes. 112 However, the combination therapy in elderly AML patients compared with induction chemotherapy alone failed to show any survival benefit and even led to increased adverse events. 113 However, several clinical trials are currently active and enrolling patients [ClinicalTrials.gov identifiers: NCT03417427, NCT01839240, NCT02275663] and the identification of predictive molecular biomarkers and changes to the administration schedules (to decrease toxicity and increase synergy) may improve outcomes of future trials.
Finally, the combination of AZA with the NEDD8-activating enzyme inhibitor pevonedistat showed synergistic effects in a phase Ib trial in R/R-AML patients. 114 Pevonedistat inhibits the proteosomal degradation of intracellular proteins leading to their cytotoxic accumulation. 115 While the data for pevonedistat in MDS patients is scarce, a recently presented abstract from an ongoing phase II trial [ClinicalTrials.gov identifier: NCT03238248] enrolling MDS and MDS/MPN-overlap patients after HMA failure appeared promising. 72 Among 21 evaluable patients, nine patients met the composite of CR, PR, and HI with one CR and four mCRs. The toxicity profile appeared manageable, with thrombocytopenia (39%), anemia (35%), leukopenia (26%), and neutropenia (22%; 13% febrile neutropenia) being the most common ⩾grade 3 adverse events. 72 Preliminary data from the frontline setting of AZA + pevonedistat compared with AZA monotherapy were recently presented and did not show a statistically significant OS benefit (HR for death: 0.70; 95% CI 0.39–1.27; p = 0.240) despite a higher rate of CR (52% versus 27%; p = 0.05) for the combination therapy arm in the subgroup of MDS patients. 116 However, additional data from ongoing studies in both the frontline [ClinicalTrials.gov identifier: NCT03268954; PANTHER trial] and relapsed setting [ClinicalTrials.gov identifier: NCT03772925] are necessary.
Future directions
While the NCCN guidelines still recommend HMA monotherapy as the first line option for many MDS patients, the convincing results of combinations of HMAs with ICI in both the frontline and HMA-refractory setting may become a valid alternative option in the future.68,85,117 Since ICI have only limited activity as monotherapy, HMAs will continue to play a key role in MDS treatment and remain as the backbone of MDS therapy in the future.
Optimization of upfront therapy will be key in addressing the optimal MDS therapy given the poor prognosis of MDS when it is refractory to HMAs. The development of effective salvage regimens is dire, and a current unmet need. For MDS patients with stable or progressive disease while on HMA, two trials have tested the addition of other agents to AZA as potential salvage regimens. However, both the addition of the HDAC inhibitor vorinostat and of the smoothened hedgehog pathway inhibitor LDE225 have had only lackluster results with salvage rates of 10% and 13%, respectively, as well as median OS of 12 months and 7 months, respectively.118,119 This suggests that treatment intensification at the time of progression might be too late and optimized frontline treatment by, for example, addition of venetoclax or ICI might be more effective. However, the promising results seen with HMA + venetoclax combination therapy in AML patients need to be confirmed in MDS patients first before this combination can be considered for routine upfront use. In addition, a better understanding of the pathophysiologic mechanisms underlying bone marrow progression and HMA failure is necessary to optimize existing and develop novel therapeutic options.
While several other agents have been, and are being, tested as monotherapies in MDS patients in the HMA-refractory setting, none of these appears to have the potential to replace HMAs.
For example, glasdegib is a smoothened inhibitor that targets the hedgehog pathway whose upregulation has been implicated in the pathogenesis of myeloid neoplasms and leukemic stem cell survival. 120 In a phase II study of 35 MDS patients refractory to HMAs, glasdegib showed an ORR of 6% (n = 2 patients) and a median OS of 10.4 months. 121 Treatment appeared to be well-tolerated, with grade 3 or higher infections and 30-day mortality in 11% of patients (n = 4) each. 121 The limited effect of glasdegib as a monotherapy suggests that glasdegib probably does not have a role as monotherapy in the treatment landscape, but response rates in combination with LDAC or AZA appear to be superior and are being investigated further.122,123
Rigosertib is a multikinase inhibitor that targets the oncogenic Ras and PI3K pathways by binding to the Ras-binding domain of various kinases.124,125 Several clinical trials have studied rigosertib in HMA-refractory patients. The largest phase III trial randomized 299 HMA-refractory, high-risk MDS patients in a 2:1 ratio to rigosertib or best supportive care. Unfortunately, median OS was similar in both groups [8.2 months (95% CI 6.1–10.1) in the rigosertib group and 5.9 months (4.1–9.3) in the best supportive care group (p = 0.33)]. 125 This is similar to a prior smaller phase I/II study that showed a median OS of 35 weeks with 40% bone marrow blast responses. 124 However, in the minority of patients who do respond to rigosertib the median OS is significantly longer and has been reported to be as high as 15.7 months. 40 Data for rigosertib in the frontline setting and/or in combination with HMA are not available yet but clinical trials are ongoing [e.g., ClinicalTrials.gov identifier: NCT01926587] and synergistic effects have been suggested by preclinical experiments. 126
One of the major changes in the treatment of MDS patients will be a more individualized approach to treatment selection thanks to advances in genetic testing. While testing for somatic mutations is not yet standard practice, 127 previous studies have shown that patients with TET2 and DNMT3A mutations may have a higher response rate to HMA therapy.128,129 Conversely, patients with ASXL1 mutations or who harbored four or more mutations had a lower likelihood of response to HMAs and an adverse OS. 130 With further advances in diagnostic techniques such as next generation sequencing (NGS), identification of targetable mutations may enable a more individualized approach to the upfront treatment for MDS patients. Besides mutations that are direct targets for already FDA-approved medications such as IDH1/2 or FLT3, the spliceosome mutation SF3B1 has been identified as a predictive biomarker for a high response rate to treatment with the TGF-β pathway inhibitor luspatercept in patients with low-risk MDS and ringed sideroblasts.131–136
As another example of molecularly targeted therapies, the orally available spliceosome inhibitor H3B-8800 has been tested in a phase I trial [ClinicalTrials.gov identifier: NCT02841540] in 84 patients with CMML, MDS, and AML, of whom 87% had received prior HMA therapy and 88% of included patients harboring mutations in SF3B1, U2AF1, SRSF2, or ZRSR2. 137 Although dose-dependent changes in the splicing pattern were seen with H3B-8800 and the treatment was shown to be safe, none of the patients in the study achieved a CR or PR. 137 However, further studies are needed to assess the full efficacy of this agent.
APR-246, a small molecule that stabilizes and restores wild-type activity of mutant p53 and induces apoptosis selectively in TP53-mutant cells, is another promising molecularly targeted novel therapy.75,138 In two separate phase I/II clinical trials the combination of AZA and APR-246 has shown synergistic effects with ORR of 75–87%, with 53–56% CR reported among TP53-mutated, HMA-naïve patients with MDS, CMML, or AML with <30% blasts.75,76 Given that TP53 mutations have been frequently associated with adverse outcomes and poor cytogenetic features such as complex karyotypes and therapy-related myeloid neoplasms, these results are especially encouraging.24,56 However, increasing evidence suggests that not all TP53 mutations carry the same prognostic relevance and a more nuanced approach might be warranted.139,140 Furthermore, these results need to be verified in the ongoing placebo-controlled, randomized phase III trial comparing AZA + APR-246 with AZA + placebo [ClinicalTrials.gov identifier: NCT03745716].
Compared with AML, targetable mutations in MDS are rare, with IDH1/2 and FLT3 mutations encountered in less than 5% of patients with MDS, and all of the currently available agents (ivosidenib, enasidenib, midostaurin, gilteritinib) have been tested in, and are FDA-approved only for, AML.23,141 Given the closely linked disease biology of AML and MDS, these agents may be considered for off-label use in the selected minority of MDS patients who harbor these mutations. Furthermore, there are limited data that support a potential role for the IDH inhibitors enasidenib and ivosidenib in HMA-refractory MDS as well. The IDH2 inhibitor enasidenib has been tested in 16 IDH2-mutated MDS patients (11 HMA-refractory patients) and showed an ORR of 53% (8/15 evaluable patients) in the entire cohort and 50% in the HMA-refractory setting. 142 Even higher response rates have been reported in a phase I study of 12 IDH1-mutated MDS patients (9 HMA-refractory) who were treated with ivosidenib. ORR in this study was 91.7% (11/12 patients), with 5 patients (41.7%) achieving CR. 143 Given the high – and in some cases durable response rates – with a favorable side effect profile, these agents could potentially be even considered as first-line alternatives to or in combination with HMAs. However, clinical trial data from MDS patients are scarce and need to be validated in additional studies. Several trials testing enasidenib [ClinicalTrials.gov identifiers: NCT03383575, NCT03744390] and ivosidenib [ClinicalTrials.gov identifier: NCT03503409] alone or in combination with AZA are currently active and will provide further information on the safety and efficacy of these agents in the treatment of MDS. While making treatment decisions based on NGS information is a rapidly evolving field, it is not quite ready for incorporation in the upfront treatments in the MDS patients at this time.
Conclusion
HMAs remain the mainstay of therapy for the majority of MDS patients. However, response rates are merely 40–50% and therapeutic effects are often only transient. Given the poor prognosis of HMA-refractory patients, additional therapies are desperately needed with combinations of HMAs and venetoclax, ICI or targeted agents appearing to be the most promising options (although not without toxicity). HMAs, especially in combination therapy, will remain the backbone of MDS treatment and some targeted therapies will likely be added for specific appropriate populations once additional safety and efficacy data are accumulated (e.g., IDH1/2, FLT3). While not ready for routine use in clinical practice, tailoring treatment concepts based on NGS testing may improve outcomes by allowing for a more targeted and individualized therapy.
Footnotes
Author contributions
JPB and TP wrote the manuscript. HEC edited the manuscript and included additional salient studies and data.
Conflict of interest statement
JPB has no conflicts of interest to declare.
TP reports research support from JAZZ, Agios, BMS, and consulting for Genentech, Tetraphase
HC reports research support from Celgene, consulting for Celgene, Agios, Sanofi, Jazz, Novartis, Stemline. IRC committee work for ABBVIE and Takeda.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the DELUCA foundation.