
Abstract
Acute myeloid leukemia (AML), the most prevalent acute leukemia in adults, is an aggressive hematological malignancy arising in hematopoietic stem and progenitor cells. With the exception of a few specific AML subtypes, the mainstays of treatment have not significantly changed over the last 20 years, and are still based on standard cytotoxic chemotherapy. As a result, clinical outcome remains poor for the majority of patients, with overall long-term survival in the region of 20–30%. Recent successes in characterizing the genetic landscape of AML have highlighted that, despite its heterogeneity, many cases of AML carry recurrent mutations in genes encoding epigenetic regulators. Transcriptional dysregulation and altered epigenetic function have therefore emerged as exciting areas in AML research and it is becoming increasingly clear that epigenetic dysfunction is central to leukemogenesis in AML. This has subsequently paved the way for the development of epigenetically targeted therapies. In this review, we will discuss the most recent advances in our understanding of the role of epigenetic dysregulation in AML pathobiology. We will particularly focus on those altered epigenetic programs that have been shown to be central to the development and maintenance of AML in preclinical models. We will discuss the recent development of therapeutics specifically targeting these key epigenetic programs in AML, describe their mechanism of action and present their current clinical development. Finally, we will discuss the opportunities presented by epigenetically targeted therapy in AML and will highlight future challenges ahead for the AML community, to ensure that these novel therapeutics are optimally translated into clinical practice and result in clinical improvement for AML patients.
Introduction
Acute myeloid leukemia (AML) is the most prevalent acute leukemia in adults and results from the transformation of primitive hematopoietic stem and progenitor cells (HSPCs), leading to increased proliferation and impaired differentiation of immature myeloid progenitors [Gilliland and Tallman, 2002]. The term AML encompasses multiple specific and clearly defined subtypes that are highly heterogeneous in terms of their genetics, biology and clinical behavior, highlighting the requirement for specific therapies for each subtype. However, despite our increased understanding of AML pathogenesis, the mainstay of treatment for the majority of AML subtypes has not significantly changed and continues to be based on standard cytotoxic chemotherapy consisting of anthracyclines and cytarabine [Vardiman et al. 2009; Roboz, 2012]. Prognosis is influenced by a combination of cytogenetic and molecular features of the disease, together with clinical characteristics and the patient’s age. Although treatment outcomes have improved over the last 20 years for certain groups of patients and specific subtypes, the overall outlook for the majority of patients remains dismal with only 20–30% of patients achieving long-term survival [Liesveld, 2012], further highlighting that standard chemotherapy has reached the ceiling of its effect and that novel therapies are urgently required.
One of the main advances in our understanding of AML pathogenesis has been the observation that transcriptional dysregulation and epigenetic dysfunction are common and recurring themes in AML, as demonstrated by the frequent occurrence of mutations in epigenetic and transcriptional regulators across several subtypes of AML [Cancer Genome Atlas Research Network, 2013] (Table 1). The term epigenetics was originally used to describe heritable changes in gene expression (phenotype) that were independent of DNA sequence alterations (genotype) [Waddington, 2012]. Lately the term has come to include the whole of chromatin biology as well as those processes which can alter gene expression without affecting DNA sequence, including the recently highlighted role of non-coding RNAs in gene expression [Berger et al. 2009]. However, in this review, we will focus on the role of posttranslational modifications (PTMs) at both the DNA and histone level, as their role in AML pathogenesis is better understood and has already led to the development of novel therapeutics which have shown great promise in preclinical models [Daigle et al. 2011; Dawson et al. 2011; Zuber et al. 2011; Schenk et al. 2012; Daigle et al. 2013; Wang et al. 2013] as will be further discussed.
Epigenetic regulators recurrently mutated in AML.

All proteins marked with an* also contain reader domain(s).
AML, acute myeloid leukemia; CBP, CREB binding protein; EZH2, enhancer of zest homolog 2; IDH, isocitrate dehydrogenase; JARID1A, Jumonji AT-rich interactive domain 1A; MLL, mixed lineage leukemia; MORF, MOZ-related factors; MOZ, monocytic leukemia zinc finger; NSD, nuclear receptor binding SET-Domain; NUP98, nucleoporin 98kDa; TET, ten-eleven translocation; TIF (NCOA2), nuclear receptor coactivator 2; UTX, ubiquitously transcribed tetratricopeptide repeat, X chromosome.
Chromatin is a composite of DNA and its protein scaffold, whose basic structure is called the nucleosome core complex. Each nucleosome is composed of two copies of each of the four histone proteins (H2A, H2B, H3, and H4), forming an octamer with DNA wrapped around it. The accessibility of the transcriptional machinery to chromatin affects the level to which a gene is actively transcribed and historically chromatin has been thought to be present in two physical states: euchromatin or heterochromatin. While euchromatin is in a relaxed state and allows binding of proteins modulating gene expression, heterochromatin is in a more compact state and less permissive to access of the transcriptional machinery, thus facilitating gene repression [Dawson et al. 2012]. PTMs of histone proteins appear to contribute to the physical state of chromatin, as they can modulate noncovalent interactions between DNA and histone proteins. For example, acetylation is thought to increase chromatin accessibility by altering the overall histone charge, thereby weakening the DNA–histone interaction. Moreover, a variety of histone modifications provide direct and indirect binding sites to either additional epigenetic regulators or components of the transcriptional machinery, thus leading to changes in gene expression levels. Several histone modifications and their putative role in chromatin biology and regulation of gene expression have been described to date, including acetylation, methylation, phosphorylation and ubiquitination [Kouzarides, 2007]. These modifications are produced by specific enzymes (the so-called ‘epigenetic writers’) and removed by other enzyme (the so-called ‘epigenetic erasers’). More recently, a third class of protein has been described that are able to recognize and bind to specific PTMs of histones and to modulate transcription and other DNA-templated processes such as replication and DNA repair. These proteins are generically termed epigenetic ‘readers’ [Dawson et al. 2012]. Thus, the ‘epigenetic code’ is the dynamic result of a multistage process laid down by epigenetic writers and erasers, leading to alterations in chromatic accessibility and modulation of gene expression and other DNA-templated processes mediated by epigenetic readers (Figure 1).
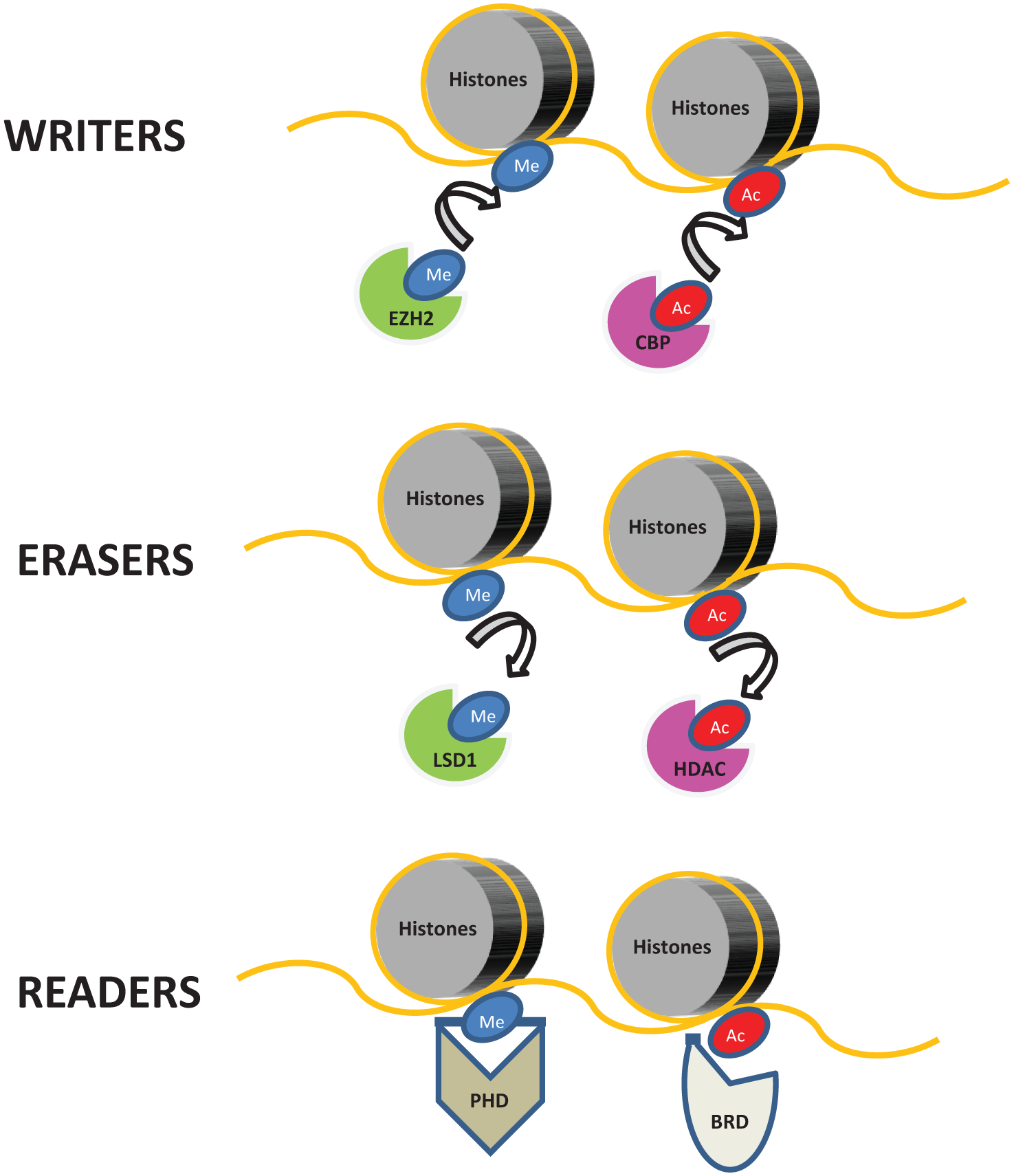
Classification of epigenetic regulators.
DNA methylation occurs at the carbon-5 position in cytosine nucleotides in the context of a 5′-cytosine-phosphate-guanine-3′ (5′-CpG-3′) dinucleotide. This process is dynamic and reflects the balance between active methylation, mediated by the two predominantly de novo DNA methyltransferases, DNMT3A and 3B, and the maintenance methyltransferase DNMT1 [Jones, 2012] and demethylation. Recently, the mechanisms of active cytosine demethylation have also been identified, with the TET (ten eleven translocation) family of DNA dioxygenases initially oxidizing cytosine through a number of intermediates (hydroxyl: hmC; formyl: fC; and carboxyl-cytosine: caC) in reactions requiring oxygen, Fe(II) and α-ketoglutarate. These intermediates are then further converted back to unmethylated cytosine through the base excision repair pathway via catalysis involving the enzyme thymidine DNA glycosylase ± AID-APOBEC (activation-induced cytidine deaminase-apolipoprotein B mRNA editing enzyme catalytic) [Abdel-Wahab and Levine, 2013]. The exact relationship between DNA methylation and transcription correlates with the position of the CpG dinucleotide within the genome, but in general, methylated cytosine (5mC), particularly in promoter CpG islands, associates with heterochromatin and with transcriptional repression [Jones, 2012]. Conversely, 5hmC is mostly associated with euchromatin and an increase in 5hmC has been documented at the transcriptional start sites of expressed pluripotent genes in embryonic stem (ES) cells [Ficz et al. 2011]. Analogous to modifications of histones, 5mC is bound by specific methylbinding proteins, presumably to translate the DNA methylation signal, and growing evidence suggests that 5hmC and possibly 5fC and 5caC, are functional intermediates that bind to a specific repertoire of proteins, although the identities of these proteins are only now becoming apparent [Takai et al. 2014].
Interestingly, a growing number of epigenetic writers and erasers acquire either activating or loss-of-function mutations in AML, leading to the hypothesis that epigenetic dysfunction is central to AML pathogenesis. Targeting these epigenetic alterations can modulate the transcriptional programs altered in AML, opening novel therapeutic windows in this aggressive cancer. Although the number of epigenetic regulators reported to be mutated/dysregulated in AML is fast growing (Table 1), in this review we will specifically focus on those epigenetic regulators that have already been demonstrated to be central to AML pathogenesis and whose therapeutic targeting has been shown to be effective in AML preclinical models, facilitating their progression into clinical trials.
IDH1/2 mutations: mechanisms leading to leukemogenesis and their therapeutic targeting
Isocitrate dehydrogenases (IDHs) are responsible for the oxidative decarboxylation of isocitrate to α-ketoglutarate. Three different isoforms of the IDH protein are present in humans. IDH3 is a nicotinamide adenine dinucleotide (NAD+)dependent isoform localized in the mitochondrial matrix which plays a central role in aerobic energy production via the Krebs cycle. In contrast, the other two isoforms (IDH1 and IDH2) are nicotinamide adenine dinucleotide phosphate (NADP+) dependent and similar to each other. They mediate other metabolic processes, including lipid metabolism and glucose sensing (IDH1) and oxidative respiration (IDH2) [Reitman and Yan, 2010]. Mutations in the genes encoding IDH1 and 2 tend not to occur together in the same clone and, collectively, have been described in 10–20% of AML, predominantly in cytogenetically normal cases [Mardis et al. 2009; Marcucci et al. 2010; Chotirat et al. 2012]. These alteration-of-function mutations result in proteins with neomorphic activity that show increased affinity for α-ketoglutarate and furthermore promote its subsequent reduction to 2 hydroxyglutarate (2-HG) [Ward et al. 2010]. 2-HG accumulates in leukemic cells where it acts as an ‘oncometabolite’ due to its ability to interfere with the functions of epigenetic modifiers with catalytic activity that requires α-ketoglutarate as an enzymatic cofactor, including the TET family of methylcytosine dioxygenases [Xu et al. 2011] and the Jumonji C (JmjC) domain-containing family of histone lysine demethylases [Chowdhury et al. 2011]. Respectively, this inhibition leads to increases in both DNA and histone methylation, thus linking mutations in metabolic enzymes to dysregulated transcriptional programs in AML [Figueroa et al. 2010; Lu et al. 2012]. It is notable in this respect that IDH1/2 and TET2 mutations are also mutually exclusive in AML and as would be predicted by their functional phenocopy of each other, patients carrying these mutations show similar DNA methylation profiles [Figueroa et al. 2010]. Although several IDH1/2 mutations have been described in AML, they have all been found to be gain-of-function mutations, strengthening their involvement in AML pathogenesis [Chotirat et al. 2012]. However, the prognostic significance of IDH1/2 mutations in AML is still unclear as contrasting results have been published, with some series showing an association with adverse outcome and others with an improved outcome [Boissel et al. 2010; Paschka et al. 2010; Thol et al. 2010; Green et al. 2011; Chotirat et al. 2012; Koszarska et al. 2013]. It is possible that their prognostic significance depends on the specific allele and the identity and combination of other compound mutations as well as patient variables such as age and fitness.
The mechanisms by which IDH1/2 mutations promote leukemogenesis have been investigated in vitro and in vivo in different models and appear to be secondary to impaired hematopoietic differentiation and expansion of stem/progenitor cells [Figueroa et al. 2010; Lu et al. 2012; Losman et al. 2013]. Hematopoietic-specific IDH1 (R132H) conditional knock-in mouse models result in marked myeloproliferation, but do not progress to overt AML, strongly suggesting that additional mutations are required for AML development. These mice display an expansion of their hematopoietic stem cells (HSCs) and myeloid progenitor (MP) compartments, both in their bone marrow (BM) and spleens. Moreover the DNA methylation signature within their HSC and MP compartments showed significant similarities with that observed in patients with AML carrying IDH1/2 mutations [Sasaki et al. 2012]. In consonance with these findings, a similar phenotype was demonstrated using a retroviral transduction/transplantation mouse model. Wild-type mouse BM cells retrovirally transduced with IDH1 mutant alleles fail to generate disease. However, ectopic overexpression of the IDH1 mutant allele in HoxA9-immortalized BM cells significantly shortens the latency of the resulting myeloproliferative-like myeloid leukemia [Chaturvedi et al. 2013]. Together, these results suggest that IDH1 mutations are key events in leukemogenesis, but additional genetic or epigenetic factors are required for AML transformation.
Regardless of their exact role in leukemogenesis, the novel function of IDH1/2 mutant proteins has provided an ideal target for leukemia-specific therapies and the development of IDH1/2 inhibitors has therefore gained significant momentum over the last few years, with many such inhibitors currently being assessed in preclinical models. HMS-101 is a specific inhibitor of the IDH1 mutated protein which has been discovered via a computational drug screen using the ZINC library [Irwin et al. 2012]. HMS-101 is active in vitro against both mouse BM cells and primary human AML cells carrying IDH1 mutations [Chaturvedi et al. 2013], and more recently has been shown to specifically inhibit 2-HG production by mutant IDH1 in vivo, while simultaneously reducing proliferation and inducing differentiation in leukemic cells. These effects translated in a prolonged survival of IDH1 mutant leukemic mice treated with HMS-101 and await further confirmation in clinical trials [Chaturvedi et al. 2014]. Compounds targeting mutant IDH2 have also been studied in leukemia cell lines and primary AML samples [Losman et al. 2013; Wang et al. 2013] and were shown to reduce 2-HG production and lead to differentiation of leukemia cells in a mutant-specific manner. These effects correlated with global changes in DNA methylation/histone state and suggest the induction of differentiation through alterations in the epigenetic state of mutant cells as a putative mechanism of action for these compounds [Kernytsky et al. 2014]. These data have led to the clinical development of AG-221, an oral, potent, reversible, and selective inhibitor of the mutant IDH2 protein. A phase I study of AG-221 [ClinicalTrials.gov identifier: NCT01915498] in patients with advanced IDH2 mutant positive hematological malignancies is currently underway (Table 2) and very encouraging preliminary results have recently been reported [Stein et al. 2014a]. AG-221 has been well tolerated and 20/32 (62.5%) of subjects evaluable for efficacy have achieved an objective response, with 11 complete responses (CRs) and 8 partial responses (PRs) documented. Moreover, five subjects have stable disease and still remain on the study drug. Responses may also be relatively durable, as complete remissions of up to 4.5 months have been observed. These responses were associated with induced differentiation of myeloblasts into mature forms, consistent with preclinical models and all responding patients demonstrated neutrophil recovery. Early results therefore recapitulate the response observed in preclinical models and further confirm that IDH inhibitors are likely to be incorporated in the specific management of patients with AML with IDH mutations.
Clinical trials of novel epigenetic therapies in AML from ClinicalTrials.gov as of December 2014.*
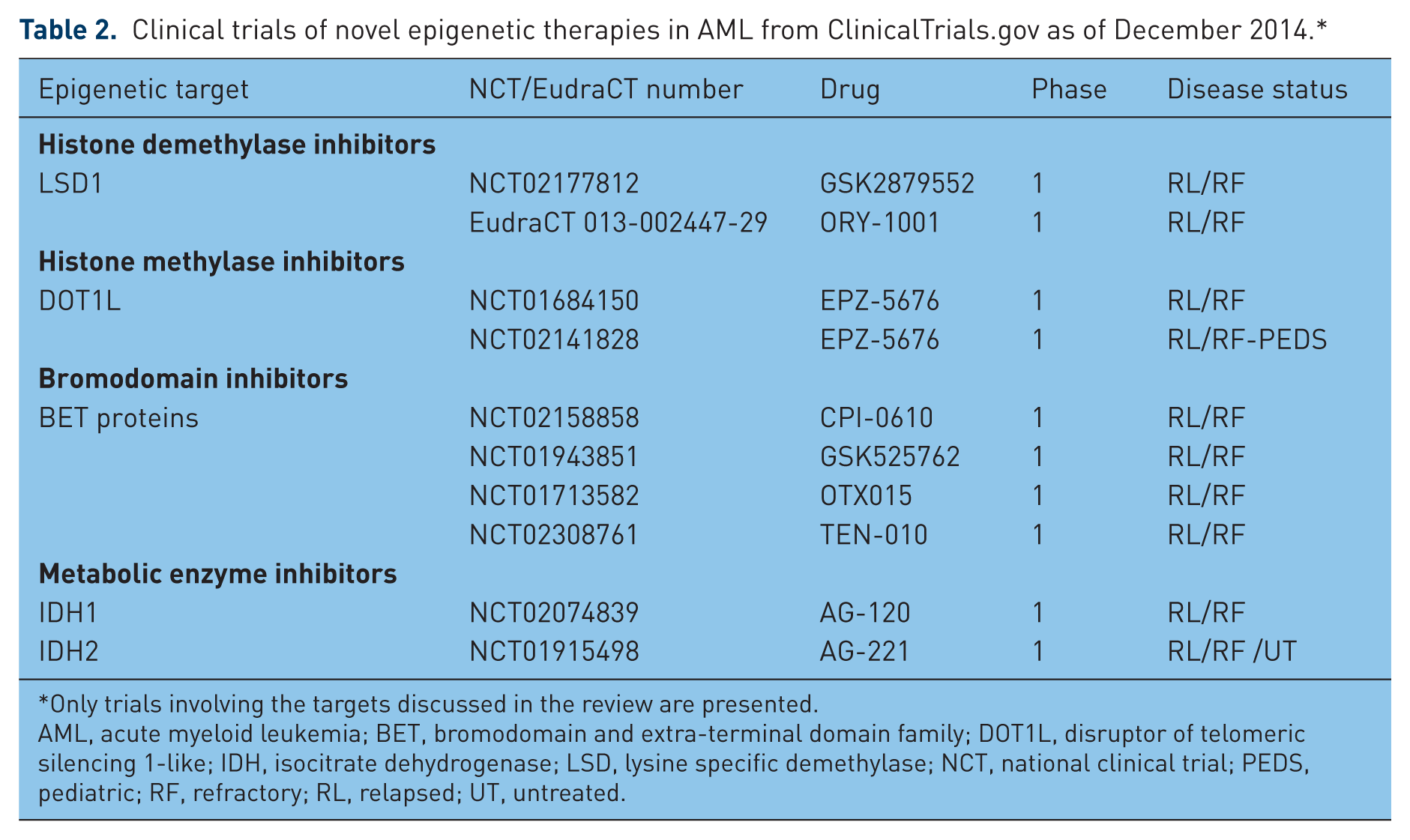
Only trials involving the targets discussed in the review are presented.
AML, acute myeloid leukemia; BET, bromodomain and extra-terminal domain family; DOT1L, disruptor of telomeric silencing 1-like; IDH, isocitrate dehydrogenase; LSD, lysine specific demethylase; NCT, national clinical trial; PEDS, pediatric; RF, refractory; RL, relapsed; UT, untreated.
Lysine specific demethylase 1: its role in leukemogenesis and as a potential therapeutic target in AML
First identified in 2004 [Shi et al. 2005], lysine specific demethylase 1 (LSD1) has emerged as a promising therapeutic target in multiple cancers, notably in AML [Berglund et al. 2008; Lokken and Zeleznik-Le, 2012; Schenk et al. 2012; Fiskus et al. 2014c; Niebel et al. 2014]. Its main role is demethylation of H3K4me1/2 and H3K9me1/2 (although it can also demethylate nonhistone proteins such as DNMT1 and TP53) and LSD1 has been shown to dynamically affect a wide range of transcriptional programs in a context-specific manner, acting either as a transcriptional repressor or activator [Metzger et al. 2005; Wang et al. 2007, 2009; Cai et al. 2011; Huang et al. 2007; Kerenyi et al. 2013]. Increased LSD1 expression has been reported in a variety of solid organ tumors, including bladder, lung and colorectal carcinomas, and also in myeloproliferative disorders, including chronic myeloid leukemia, myelodysplastic syndromes, and multiple subtypes of AML [Berglund et al. 2008; Hayami et al. 2011; Niebel et al. 2014]. LSD1 had been shown to be a component of a mixed lineage leukemia (MLL) super complex associated with sites of active transcription [Nakamura et al. 2002], which led to the hypothesis that it might have a direct and functional role in MLL-driven leukemogenesis. Indeed, analysis of microarray data from 23 murine MLL leukemias initially revealed a significant correlation between LSD1 expression levels and clonogenic potential [a surrogate for leukemic stem cell (LSC) frequency] in methylcellulose-based plating assays [Harris et al. 2012]. LSD1 knockdown was shown to induce terminal differentiation of AML cell lines in vitro and resulted in significant decreases of clonogenic potential across multiple human cell lines, murine leukemias and primary samples with an MLL rearrangement. Similar results were obtained in vivo, where LSD1 knockdown impaired LSC numbers and function, as evidenced by reduced engraftment efficiency in secondary recipient mice. Interestingly, the levels of global H3K4me2 remained unchanged upon LSD1 inhibition. However, further ChIP-seq analysis of MLL-AF9 murine tumors suggested that overexpression (or ectopic expression) of LSD1 may exert its oncogenic function by affecting the H3K4me2 status of specific loci bound by the MLL-AF9 fusion. Thus, sh-RNA knockdown, or pharmacological inhibition of LSD1, can directly corrupt the oncogenic program of MLL-AF9, whilst largely sparing global levels of H3K4me2. Similar findings have also been reported in a non-MLL leukemia context, when pharmacological inhibition of LSD1 was able to sensitize LSCs towards the prodifferentiation effects of all-trans-retinoic acid (ATRA) treatment, irrespective of promyelocytic leukemia gene-retinoic acid receptor-alpha (PML-RARA) status [Schenk et al. 2012]. Despite the great promise that these preclinical studies have shown, the possible side effects of LSD1 inhibition in normal hematopoiesis have not been conclusively established. In one of these studies, treatment with LSD1 inhibitors at the required antileukemic doses resulted in severe anemia and impaired erythropoiesis [Harris et al. 2012]. This study and others have also shown that LSD1 loss greatly impairs normal granulopoiesis, erythropoiesis and platelet production [Harris et al. 2012; Sprussel et al. 2012]. However, these effects were transient and reversible upon treatment discontinuation. The data from these preclinical studies strongly suggest that the efficacy of LSD1 inhibition as an AML treatment will be largely dependent on the subtype (e.g. MLL status) and may offer maximum benefits when used in combination with other compounds such as ATRA. Two phase I studies using compound GSK2879552 [ClinicalTrials.gov identifier: NCT02177812] and ORY-1001 (EudraCT 2013-002447-29) in patients with AML are currently underway (Table 2) and will address these questions.
Disruptor of telomeric silencing 1 like: its role in leukemogenesis and as a potential therapeutic target in AML
Disruptor of telomeric silencing 1 like (DOT1L) is the only histone H3 lysine 79 (H3K79) methyltransferase in mammals. It was originally identified in yeast where its overexpression led to disruption of telomeric silencing, hence its name [Singer et al. 1998]. The H3K79 methylation mark is normally associated with active transcription and H3K79 methylation has been proposed to be a critical histone modification regulating cell proliferation, as its genetic silencing leads to abnormal mitotic spindle formation and cell cycle arrest at the G1 phase [Kim et al. 2012b]. DOT1L is known to interact with the phosphorylated C-terminal domain of actively transcribing RNA polymerase II (RNAPII) and through this interaction, DOT1L and subsequent H3K79 methylations are targeted to actively transcribed genes [Kim et al. 2012a]. Beside its effects on cell cycle, DOT1L appears to also play a role in DNA repair as methylated H3K79 serves as a docking site for the global genomic repair machinery [Tatum and Li, 2011]. A crucial role for DOT1L during embryonic development has been demonstrated using germline Dot1l knockout mouse models. These embryos die between E10.5 and E13.5 from a complete absence of erythropoiesis [Feng et al. 2010]. The role of Dot1l during later hematopoiesis is however less clear, as different conditional knockout models have been used. Ubiquitous deletion of Dot1l in 6–10-week-old mice using a ROSA26-Cre-ER recombinase leads to death secondary to severe anemia and BM hypocellularity [Jo et al. 2011] and consistent findings have been published using a very similar model [Nguyen et al. 2011a]. However, conditional excision of Dot1l in the hematopoietic compartment with the Vav-Cre recombinase leads to a moderate to severe reduction in both red and white blood cells, but does not completely abrogate multilineage hematopoiesis, suggesting that DOT1L is not an absolute requirement for adult hematopoiesis [Bernt et al. 2011]. This is critical from a therapeutic perspective, as transformation of murine BM by MLL-fusion oncogenes was demonstrated to be absolutely dependent upon the presence of DOT1L [Chang et al. 2010; Jo et al. 2011; Nguyen et al. 2011b]. Taken together, these data suggest a differential therapeutic window for inhibition of DOT1L that could be exploited in the management of certain subtypes of AML. Moreover, recent studies confirm that MLL-driven leukemias (MLL-AF9 and MLL-AF6) display abnormal H3K79me2 patterns on direct target genes of the MLL-fusion proteins which are distinctive compared with other transcriptionally active loci within the same cell and to the same loci when expressed under physiological conditions during normal hematopoiesis [Bernt et al. 2011; Deshpande et al. 2014]. This specific epigenetic mark of MLL-fusion targets might explain the nonphysiological dependence of the MLL-fusion driven leukemogenic transcription program on H3K79 methylation and DOT1L. In support of this model, only those MLL-fusion target genes which are central to the transformation ability of MLL oncogenes, such as the HoxA cluster and the homeobox gene Meis1, were shown to be downregulated following loss of DOT1L and H3K79 methylation [Bernt et al. 2011; Nguyen et al. 2011b]. These specific effects might be secondary to the fact that DOT1L directly interacts with several known partners of MLL fusions [Okada et al. 2005; Zhang et al. 2006; Mueller et al. 2007], particularly the AF10 protein, and is therefore aberrantly recruited to gene targets of these MLL fusions. Taken together, these studies provide support for a critical role of DOT1L in leukemogenesis driven by MLL fusions and its therapeutic targeting by small molecule inhibitors.
Several preclinical studies have been reported using DOT1L inhibitors which compete with S-adenosyl methionine, the methyl donor required for the methyltransferase activity of DOT1L [Daigle et al. 2011, 2013]. These studies have shown that the inhibitors specifically reduce H3K79 methylation marks in leukemic cells together with a reduction in the expression of MLL-fusion target genes (Figure 2). These effects are linked to similar effects on proliferation and viability of MLL-rearranged leukemias and similar effects were also reported in xenograft models of MLL leukemias in vivo. Recently, preclinical models of AML associated with another leukemogenic fusion, NUP98-NSD1, have also been shown to be sensitive to small molecule inhibition with DOT1L inhibitors, suggesting more widespread roles in AML therapy [Deshpande et al. 2014]. Moreover, toxic effects were not observed on healthy mice, thus suggesting that DOT1L chemical inhibition might be better tolerated than gene deletion, possibly as a result of incomplete inhibition, increased sensitivity of leukemic cells or methyltransferase-independent functions of DOT1L that are not targeted by the inhibitors.

Model for the mechanism of action of DOT1L inhibitors.
EPZ-5676, the most promising of all the DOT1L inhibitors tested in preclinical models, has now entered phase I clinical trials in patients with advanced hematological malignancies, including AML with MLL fusions [ClinicalTrials.gov identifier: NCT01684150] (Table 2). Although the compound has some pharmacokinetic limitations, requiring continuous infusions of up to 28 days, it appears to be well tolerated and was shown to lead to inhibition of H3K79 methylation. CRs have been observed in a small number of patients and continued clinical investigation is currently ongoing [Stein et al. 2014b].
Targeting BRD4 and other bromodomain and extra-terminal proteins in AML
The bromodomain and extra-terminal (BET) protein family of epigenetic readers are transcriptional adapter molecules that, in general, facilitate transcription [Florence and Faller, 2001; Zeng and Zhou, 2002; Wu and Chiang, 2007]. They comprise bromodomain-containing protein (BRD)2, BRD3, and BRD4, which are ubiquitously expressed and also BRDT, whose expression is confined to the testes [Pivot-Pajot et al. 2003; Wu and Chiang, 2007]. As their name would suggest, they contain tandem N-terminal bromodomains, specialized epigenetic reader modules that bind to acetylated lysine residues of histone (and nonhistone) proteins, mediating effects that can range from histone modifications to chromatin remodeling and ultimately leading to transcriptional activation [Taverna et al. 2007]. BET proteins are essential for cellular homeostasis, as evidenced by knockout mouse models demonstrating embryonic lethality [Houzelstein et al. 2002; Dey et al. 2003; Kanno et al. 2004; Wu and Chiang, 2007; Shang et al. 2009]. Recently, they have also been implicated in transcriptional dysregulation in many cancer types, with BRD4 identified as a key player in AML [Dawson et al. 2011, 2014; Delmore et al. 2011; Mertz et al. 2011; Zuber et al. 2011; Herrmann et al. 2012; Loven et al. 2013; Stewart et al. 2013; Fiskus et al. 2014a]. BRD4 and other BET proteins have been shown to activate transcription through facilitating transcriptional initiation via binding to transcription factors such as nuclear factor κB [Yang et al. 2005; Zou et al. 2014] and also by their recruitment of the positive transcription elongation factor b (P-TEFb) complex that facilitates release of RNAPII from proximal promoter pausing and transcriptional elongation (Figure 3). We and others have also demonstrated the binding of BET proteins to the ‘so-called’ super elongation complex and the polymerase-associated factor complex, two complexes integral to the transforming ability of MLL-rearranged leukemias [Dawson et al. 2011, 2012]. BRD4 has also been identified as an important modulator of oncogene expression in multiple myeloma and non-Hodgkin lymphoma through similar transcriptional dysregulation [Loven et al. 2013].

Model for the mechanism of action of bromodomain and extra-terminal (BET) protein inhibitors.
In addition to their role in the control of transcriptional initiation and elongation, recent studies have also implicated BRD4 and BET proteins in the regulation of enhancer function, particularly large enhancers (commonly called stretch or super enhancers) that control a number of developmentally important genes, including oncogenes [Loven et al. 2013; Dawson et al. 2014]. From studies in cell lines and patient samples, a number of genes critical for the maintenance of the leukemic phenotype, including BCL-2, IRF8, and c-MYC, have been identified as BET/BRD4 dependent and their downregulation upon inhibition with either the JQ1 or I-BET small molecule BET inhibitors leads to cell cycle arrest and apoptosis. Furthermore a significant survival advantage has been demonstrated for treated animals in murine models of aggressive MLL-rearranged leukemias [Dawson et al. 2011; Zuber et al. 2011]. Although initial efficacy and mechanism was demonstrated in MLL-rearranged leukemias, small molecule BET inhibitors have since been used successfully in in vitro and in vivo studies against a wide range of non-MLL-driven AMLs, including those that carry mutations in nucleophosmin (NPM1), FLT3-ITD and DNMT3A genes [Stewart et al. 2013; Dawson et al. 2014; Fiskus et al. 2014a], strengthening the notion that c-MYC inhibition is not the sole mechanism through which these compounds exert their antitumor potency. For NPM1c mutant AML, our own data predict that mutation of NPM1 may relieve an inhibitory interaction between wild-type NPM1 and BRD4, through the known cytosolic dislocation of both NPM1c and, via heterodimerization, wild type NPM1. This would then allow BRD4 to activate aberrant transcription at critical loci in NPM1c AML, explaining the sensitivity of this subtype to BET inhibition [Dawson et al. 2014]. However, for the majority of other AML subtypes, the exact mechanisms of action remain to be elucidated, as does the relative contribution of inhibition of transcriptional initiation, elongation, and enhancer function to overall BET inhibitory function. Most importantly, BRD4 expression can be seen both in the cytoplasm and in the nuclei of LSCs coming from multiple AML primary samples (as well as AML cell lines), irrespective of the subtype or disease stage [Herrmann et al. 2012], suggesting that BRD4 may be directly involved in the transcriptional programs that control LSC potential, and thus, its inhibition may target the population that is mainly responsible for disease dissemination.
Taken together with promising results from combination treatment studies in which BET inhibitors were used together with FLT3 tyrosine kinase inhibitors [Fiskus et al. 2014a], conventional cytostatic compounds (e.g. ARA-C) [Herrmann et al. 2012] and histone deacetylase inhibitors [Fiskus et al. 2014b], inhibition of BRD4/BET proteins has emerged as a therapeutic option in the future of AML treatment. This has led to the introduction of a number of early phase dose escalation safety studies using different BET inhibitors that are currently underway (Table 2). The most mature of these, utilizing the OTX015 inhibitor [ClinicalTrials.gov identifier: NCT01713582], recently reported no dose-limiting toxicities in 28 patients at or below the maximum tolerated dose of 120 mg daily [Dombret et al. 2014]. Of these patients, and with short follow up, five patients (18%) have demonstrated a clinically relevant response, with a sustained CR, one CR with incomplete platelet resolution and three PRs. Interestingly, two of the five patients had an NPM1c mutation and four of the five patients poor risk characteristics (secondary or therapy-related AML). Further clinical evaluation of BET inhibitors is ongoing and the results are eagerly awaited.
Conclusion: opportunities and challenges ahead for AML epigenetic therapeutics
Altered epigenetic regulation is a recurrent theme in AML and over the last 5–10 years a greater understanding of the processes underlying this has led to the identification of multiple rational therapeutic targets. Whilst these have provided new opportunities for the management of this unmet clinical problem, as evidenced by the evaluation of multiple epigenetic inhibitors in clinical trials, several questions remain to be answered before we can fully optimize and realize the promise of epigenetic therapies. The most prosaic of these are to recapitulate the safety and efficacy observed in preclinical studies in rigorous clinical trials. Another obvious question for these and indeed for all other AML therapies is how to identify and target the disease initiating and propagating compartment in each patient. The clonal architecture and hierarchy in individual patients with AML is extremely complex and dynamic and has been described as a ‘moving target’ due to clonal evolution. In view of this the use of different agents, potentially at different times, may be required to eradicate both the founding clone and all of its subclones, thereby achieving cure [Ding et al. 2012]. Moreover, genetically defined tumor subclones have been shown to possess unique phenotypic or functional properties that reflect some aspects of a tumor’s history and that may be able to predict its potential for relapse or resistance to therapy [Klco et al. 2014]. More recently, preleukemic HSCs have been formally demonstrated in several AML patient samples. These cells are resistant to induction chemotherapy, persist at remission, and presumably act as a reservoir from which relapse arises [Shlush et al. 2014]. These studies highlight further the need to target all clones in patients with AML, with a particular focus possibly on those that may directly contribute to disease resistance or relapse, and accordingly the design of single agent and combinatorial trials must address effects on LSCs specifically.
Many potentially useful agents are withdrawn from further development based on a lack of efficacy in early phase trials performed in patients who have relapsed, a patient group that is molecularly and cytogenetically heterogeneous and complex, as well as clinically very difficult to manage. In particular, epigenetic therapies, whose tempo of effect may be slower (cf. the demethylating agent azacytidine) [Fenaux et al. 2009, 2010] and that, due to their more targeted approach, may show efficacy only in specific AML subtypes, may be especially vulnerable to this false-negative assumption. Therefore, testing in newly diagnosed patients might be necessary to fully assess the efficacy of novel therapeutics, especially when these drugs have demonstrated minimal toxicity to normal hematopoiesis. Furthermore, the development of specific molecular and cellular predictive response biomarkers can help in identifying the appropriate patient groups in which to use specific therapies upfront, maximizing their therapeutic response through their use in patients more likely to obtain clinical benefit. Therefore, as already applied in IDH1/2 trials, trial design should specifically target a clearly defined patient population that is most likely to benefit, based on the inhibitor’s mode of action. For other inhibitors with more pleiotropic effects (i.e. bromodomain inhibitors), when these inhibitors have demonstrated potency across a range of AML subtypes, this personalized design is less important and trial entry should be more inclusive, although as it is also likely that some AML subtypes/genotypes will be more sensitive than others, trial design and analysis should take account of this.
Epigenetic therapies are unlikely to be effective in AML as single agents, therefore it is likely that they will need to be rationally combined with other therapies to achieve their maximal effect. Therefore, clarification of the timing, schedule and order of administration with combination partners for these therapies is extremely important. These combinations should be dictated by their mechanism of action, interactions, and risk of combined toxicities with other standard and targeted therapies. Combination with a standard cytotoxic therapy at induction seems reasonable, as has successfully been used for breakpoint cluster region-Abelson (BCR-ABL) inhibitors in Philadelphia chromosome+ ALL [Fielding et al. 2014]. However, epigenetically targeted therapies, which often lack a significant cytotoxic effect on their own but might have cotoxicities with standard chemotherapy agents, might be particularly appealing as a prolonged treatment in the post-remission setting where they could target specific subclones once disease debulking has been achieved by the initial wave of standard cytotoxic treatment. This setting might also suit their potentially slower tempo of effect as alluded to previously. Although the majority of current trials focus on single-agent treatment, there is a growing body of data from preclinical and mechanistic studies to predict which combinations are worth formally testing in patients with AML. Finally, as the effects of epigenetic therapies are often seen over a prolonged period of time, novel biomarkers of response and schedules for monitoring, which could include evaluation of specific epigenetic marks, gene expression signatures and other biomarker levels, are likely to be needed to fully guide and assess the efficacy of these novel therapeutics.
In conclusion, we are witnessing an exponential growth in our understanding of the role of altered epigenetics in AML biology. This in turn has led to the development of many novel therapeutics that suggest a step change in clinical practice. It is not unreasonable to predict that specific targeted epigenetic therapies will soon become available to individual patients, in a tailored-therapy approach, chosen in a bespoke manner based on their specific disease features. In addition, it is becoming increasingly probable that some of these novel agents will be utilized in the management of other hematological malignancies such as myelodysplastic syndromes and myeloproliferative disorders, as preclinical evidence of their efficacy in these conditions has been already demonstrated [Wyspianska et al. 2014]. However, many challenges, such as further deconvoluting the complexity of a highly heterogeneous disease like AML, identifying and targeting the disease propagating reservoirs, as well as improving our ability to translate in meaningful clinical trials all the advances made in preclinical models, still need to be addressed and further investigated before these promising therapies lead to any significant clinical improvements for patients with AML and other hematological malignancies.
Footnotes
Acknowledgements
We would like to thank all the members of the Huntly Laboratory and our funders Leukaemia Lymphoma Research, Kay Kendall Leukaemia fund, the Medical Research Council UK, the Wellcome Trust, the Cambridge NIHR Biomedical Research Centre, Leukemia & Lymphoma Society US, the Academy of Medical Sciences UK and Lady Tata Memorial Trust. We apologize to writers whose work we have failed to cite due to space constraints.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.