
Abstract
Polycythemia vera (PV) and essential thrombocythemia (ET) are both classic, relatively indolent, chronic Philadelphia-chromosome-negative (Ph−) myeloproliferative neoplasms (MPNs) characterized by elevated blood counts, thrombotic as well as hemorrhagic tendencies, a variety of symptoms, cumulative risks of progression to myelofibrosis and transformation to acute myeloid leukemia over time, and long survival. Molecularly, PV is more homogenous, being driven by JAK2 mutations in virtually all cases, while ET can be JAK2-, CALR-, or MPL-mutated, as well as ‘triple negative’. Recent targeted next-generation sequencing efforts have identified other, nondriver gene mutations, some with prognostic relevance. Prevention of thrombotic and hemorrhagic complications continues to be the major focus of management, although symptoms are increasingly being recognized as a relatively unmet need, particularly in ET. Thrombotic risk stratification in PV is still based on age and history of thrombosis, while in ET, the additional contribution of JAK2 V617F to thrombotic risk is now well established. The associations of leukocytosis with clotting risk (in both conditions) and mortality (in PV) have drawn increased attention with the availability of ruxolitinib as a second-line treatment in PV. Similarly, there is a renewed interest in interferons with the emergence of ropeginterferon alfa-2b as a potential new frontline treatment option in PV. Drug development is more difficult in ET, the most indolent of the classic Ph− MPNs, but ruxolitinib is being studied. Triggering apoptosis via the p53 pathway through pharmacologic inhibition of human double minute 2 (and synergism with interferon) is a new, promising therapeutic strategy.
Keywords
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), while morphologically distinct, are both relatively indolent, chronic myeloproliferative neoplasms (MPNs) characterized by prolonged survival and substantial risks of thrombosis and bleeding. The prevalence of PV and ET has been estimated to be 44–57 and 38–57 per 100,000 of the United States (US) population, respectively. 1 Morphologically, PV is characterized by pancytosis, panmyelosis and pleiomorphic megakaryocytes, while ET is characterized by thrombocytosis and increased numbers of enlarged, mature megakaryocytes with hyperlobulated nuclei; 2 however, with the advent of the new, lower hemoglobin and hematocrit thresholds for diagnosing PV in the 2016 World Health Organization (WHO) classification, there could be some conflation of PV, a virtually exclusively Janus kinase (JAK) 2-driven disease, and JAK2-mutated ET. The driver mutation spectrum of ET is more diverse, with approximately similar proportions of patients carrying activating mutations/indels in JAK2, MPL (the gene encoding the thrombopoietin receptor) and calreticulin (CALR) as in primary myelofibrosis (PMF), with the rest having so-called ‘triple negative’ disease. Survival in ET is superior to that in PV and may be no different than that of the age- and sex-matched healthy population. 3 While both conditions can progress to myelofibrosis (MF) and transform to acute myeloid leukemia (AML), these rates are, in general, low and lower for ET than for PV. 4 Recent efforts to genomically classify the Philadelphia-chromosome-negative (Ph−) MPN have considered both PV and ET together as ‘chronic-phase’ MPNs. 5 In the absence of agents proven to modify the natural history of these diseases and prevent progression to advanced phases, management of both conditions has primarily focused on minimizing the risks of thrombosis and hemorrhage. 6
Updates in diagnosis of PV
Major changes to the diagnostic criteria for PV were made in the 2016 revision to the WHO classification of MPNs and AML. 2 The hemoglobin and hematocrit thresholds for diagnosis were lowered to 16.5 g/dl/49% and 16 g/dl/48% for men and women, respectively, and bone marrow biopsy was made mandatory for diagnosis (except in cases fulfilling the 2008 WHO criteria of hemoglobin >18.5 g/dl in men and >16.5 g/dl in women, presence of a JAK2 mutation and a subnormal erythropoietin level). The main rationale behind these changes was the recognition that individuals with so-called ‘masked PV’ have inferior outcomes,7,8 possibly due to missed or delayed diagnoses and thereby, a lower intensity of treatment. 9 Given the new lower cutoffs for hemoglobin/hematocrit, bone marrow biopsy may also be helpful to distinguish between PV and JAK2-mutated ET. 10 Finally, bone marrow biopsy continues to be recommended, even in patients fulfilling the 2008 WHO criteria, to enable assessment of the presence of fibrosis at diagnosis of PV, as this has been shown to predict a more rapid progression to post-PV MF. 2
Updates in prognostication
In a multicenter study of 1545 patients with WHO 2008-defined PV, median survival for the entire cohort was projected at 18.9 years, with a trend towards worse survival than the age- and sex-matched US population (p = 0.104). 11 However, when the analysis was restricted to the 337 patients with the most mature survival data (i.e. those seen at the Mayo Clinic, Rochester, MN, USA), the median survival was only 14.1 years. Older age, leukocytosis, venous thrombosis and abnormal karyotype emerged on multivariable analysis as factors adversely impacting survival in this study. A prognostic model incorporating the first three of the above prognostic factors was developed, allotting 5 points to age ⩾67 years, 2 points to age 57–66 years, and 1 point each to leukocytosis ⩾15 × 109/l and venous thrombosis, resulting in three well-demarcated risk groups: low risk (0 points, median survival 27.8 years), intermediate risk (1–2 points, median survival 18.9 years) and high risk (⩾3 points, median survival 10.9 years). An Italian study of 435 consecutive patients with ET with 4304 person-years of follow up did not show an impact of the diagnosis on survival. 3 A history of thrombosis and male sex were independent predictors of death. A more recent study from the Mayo Clinic found the median survival of patients with ET (n = 292) to be 19.8 years, inferior to that of the age- and sex-matched US population, but better than in patients with PV (13.5 years, n= 267) and unaffected by driver mutation status. 12 That patients with prefibrotic PMF have an inferior prognosis compared with those with true ET is now well recognized, and underscores the need for careful pathologic distinction between these two entities. 13 The International Prognostic Score for ET (IPSET), based on a study of 867 patients, incorporates age ⩾60 years (2 points), prior thrombosis history (1 point) and leukocyte count ⩾11 × 109/l (1 point) into a risk model that predicts survival in WHO-defined ET; median survival was not reached in low-risk patients (0 points, n = 342), 24.5 years in intermediate-risk patients (1–2 points, n = 374) and 14.7 years in high-risk patients (3–4 points, n = 151). 14 In a very recent publication, researchers at the Mayo Clinic reported worse survival for patients with PV (median, 15 years) than those with ET (median, 18 years, p < 0.05), but similar leukemia-free survival (p = 0.22). 15 Interestingly, in this large study (665 PV, 1076 ET, 1282 PMF), patients with ET diagnosed after 1990 fared worse in terms of overall survival (p < 0.001). The risk of progression to MF was higher in patients with PV than in those with ET (p < 0.001), probably a function of the higher JAK2 V617F allele burden in PV.5,16 Of note, young patients (⩽40 years) with both PV and ET have excellent outcomes (median survival 35 + years).17,18
A targeted deep sequencing effort at the Mayo Clinic in 133 patients with PV and 183 with ET revealed one or more mutations/sequence variants in nondriver genes in 53% of patients in each cohort, the most frequent being TET2 and ASXL1, 19 both genes commonly implicated in clonal hematopoiesis of indeterminate potential.20,21 The investigators identified mutations/variants in ASXL1, SRSF2 and IDH2 in PV and those in SH2B3, SF3B1, U2AF1, TP53, IDH2 and EZH2 in ET as ‘adverse’, based on age-adjusted multivariable analysis of the impact on overall, leukemia-free or MF-free survival. In both the PV and ET cohorts, the combined frequency of these ‘adverse’ mutations/variants was 15%, and their presence was associated with inferior survival in both PV (median, 7.7 versus 16.9 years) and ET (median, 9 versus 22 years), findings that were validated in 215 Italian patients with PV and 174 with ET. 19 Building upon this work, they went on to incorporate mutational information into two new prognostic models, the Mutation-Enhanced International Prognostic Scoring Systems (MIPSS) for PV and for ET. 22 These models were derived from the study of 906 molecularly annotated patients (404 PV and 502 ET) from the Mayo Clinic (n = 416) and the University of Florence (n = 490). Median follow-up duration was approximately 10 years for the Mayo Clinic patients and 12–13 years for the Italian patients. The following factors adversely impacting survival emerged on multivariable analysis performed on all patients: in PV, age >60 years (2 points), SRSF2 mutation (2 points) and leukocyte count ⩾11 × 109/l (1 point) and, in ET, age >60 years (4 points), male sex (1 point) and SRSF2/SF3B1 mutations (2 points). The resultant four-tiered MIPSS-PV and MIPSS-ET models stratified patients into low (median survival 25.3 years in PV and 33.2 years in ET), intermediate-1 (median survival 18 years in PV and 26.3 years in ET), intermediate-2 (median survival 10 years in PV and 14 years in ET) and high-risk (median survival 5.4 years in PV and 9.4 years in ET) categories. 22 While interesting, these data do not inform management of patients with PV or ET at present, and multigene profiling of patients with PV and ET is not routine at most centers and remains largely a research tool. The vast majority of patients can expect a protracted, relatively indolent disease course with low lifetime risks of progression to MF or transformation to AML.
Investigators from multiple European centers recently reported a comprehensive genomic analysis of 2035 patients with MPN, of whom 1321 had ET and 356 had PV. They sequenced the full coding regions of 69 genes, as well as annotated single nucleotide polymorphisms for copy number profiling and germline loci that have been associated with MPN. Overall, eight mutually exclusive genomic subgroups of MPN emerged from this effort: TP53-mutated cases, cases associated with mutations in 1 or more of 16 myeloid cancer-associated genes (mostly those encoding epigenetic modifiers and spliceosome components), CALR-mutated cases, MPL-mutated cases, a JAK2-homozygous group, a JAK2-heterozygous group, cases with mutations in genes such as TET2 or DNMT3A that are not disease-specific or in genes that are typically mutated in other myeloid malignancies (for example, KIT in systemic mastocytosis), and one with no identifiable driver mutations. They developed a model for personalized prognosis (available at: https://cancer.sanger.ac.uk/mpn-multistage/) that outperformed conventional risk stratification models used for PMF, as well as the IPSET model for ET.
Updates in risk stratification for thrombosis: a role for leukocytosis?
As alluded to above, the major focus of management of both PV and ET is on the prevention of thrombohemorrhagic events. 6 In PV, age ⩾ 60 years and a history of thrombosis have been and continue to be the two main factors used to determine risk of thrombosis, which is higher than in ET, and patients with one or both of these attributes are classified as being ‘high risk’, while those with neither risk factor are considered ‘low risk’ and typically managed with phlebotomy and aspirin alone. The seminal Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) study established <45% as the hematocrit goal for all patients with PV, although the therapeutic modalities used to achieve this were not specified. 23 A subsequent multivariable, time-dependent subanalysis of the CYTO-PV data in which patients with thrombotic events were divided into approximate quartiles based on their leukocyte counts recorded at their last clinic visit before the thrombotic events occurred showed that the risk of thrombosis began to increase above a leukocyte count of 7 × 109/l, becoming statistically significant above 11 × 109/l. 24 A very similar analysis of a cohort of 1565 patients with PV from US Veterans Affairs hospitals demonstrated that patients with leukocyte counts ⩾8.5–<11 × 109/l had a significantly increased risk of thrombotic events compared with those with leukocytes <7 × 109/l, with the hazard ratio highest for those with leukocyte counts ⩾11 × 109/l. 25 Similar findings were also reported in a time-dependent analysis of the ECLAP (European Collaboration on Low-Dose Aspirin in Polycythemia Vera) study which found, after adjusting for potential confounding factors, that patients with leukocyte counts >15 × 109/l had a significantly higher risk of thrombosis compared with those with leukocyte counts <10 × 109/l. 26 Consensus guidelines from the National Comprehensive Cancer Network (NCCN) in the US recognize progressive leukocytosis, along with new thrombosis or disease-related major bleeding, progressive disease-related symptoms, a frequent or persistent need for phlebotomy with poor tolerance of the same, symptomatic or progressive splenomegaly and symptomatic thrombocytosis as potential indications for initiation of cytoreductive therapy in ‘low-risk’ patients with PV. 27
Thrombotic risk stratification in ET considers driver mutation status in addition to age and prior thrombosis history. Overall, four risk categories are recognized in the revised IPSET-thrombosis model (see Table 1): very low risk, comprising patients up to 60 years of age with no thrombosis history and wild type for JAK2, low risk, comprising those ⩽60 years and without a history of thrombosis but with JAK2 V617F, intermediate risk, consisting of individuals over 60 years with wild type JAK2 and no history of thrombosis, and high risk, defined by either a history of thrombosis (any age) or age >60 years with JAK2 V617F. 28 CALR-mutated patients with ET tend to be younger and have higher platelet counts, much lower thrombotic risk and lower hemoglobin levels and leukocyte counts than their JAK2-mutated counterparts, 29 and CALR mutation status does not modify the above thrombotic risk stratification model for ET. 30 Some data suggest that patients with ET and type 1/type 1-like CALR mutations may have a higher risk of progression to post-ET MF. 31 Because of their very low risk of thrombosis, young patients with CALR-mutated ET and no prior history of thrombosis or cardiovascular risk factors may forego aspirin: in one study, the risk of bleeding due to aspirin outweighed any benefits in terms of thrombosis prevention. 32 Twice-daily aspirin has been suggested as an alternative to cytoreductive therapy in patients at an intermediate risk, as well as in patients at a low risk with cardiovascular risk factors, 28 but this is based only on preclinical data. 33 Like in PV, leukocytosis, but not thrombocytosis, has been implicated as a risk factor for thrombosis in ET. In an analysis of 776 JAK2-annotated patients from the PT-1 study, abnormal platelet counts during follow up (but not at diagnosis) were found to be significantly associated with an immediate risk of major hemorrhage but not thrombosis, whereas elevated leukocyte counts over time significantly correlated with both thrombosis and major bleeding. 34 In another study of 891 patients with WHO-defined ET followed for a median of 6.2 years, leukocytosis (>11 × 109/l), along with age over 60 years, thrombosis history, the presence of cardiovascular risk factors and of JAK2 V617F predicted for arterial thrombosis, while a platelet count >1000 × 109/l was associated with a lower risk of arterial thrombosis; however, leukocytosis did lose significance when the analysis was restricted to JAK2-mutated patients. 35 NCCN guidelines endorse the consideration of cytoreductive therapy in patients at very low, low and intermediate risk with ET who have progressive leukocytosis, new thrombosis, acquired von Willebrand’s disease (aVWD), disease-related major bleeding, symptomatic or progressive splenomegaly, progressive disease-related symptoms, symptomatic thrombocytosis or vasomotor/microvascular disturbances not responsive to aspirin. 27
The revised IPSET model for thrombotic risk stratification in ET. 28
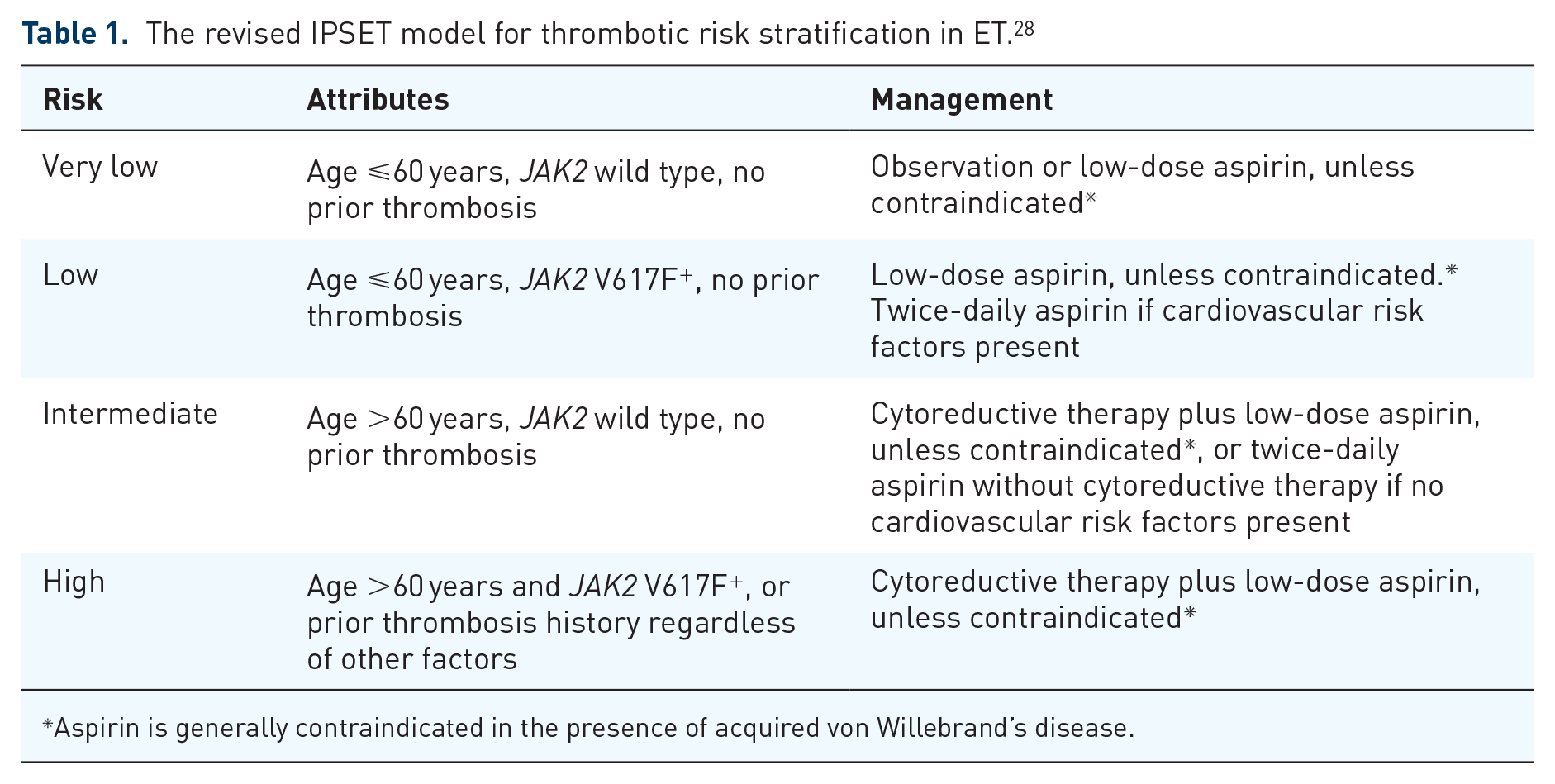
Aspirin is generally contraindicated in the presence of acquired von Willebrand’s disease.
Updates in therapy
Cytoreductive therapy is indicated for all high-risk patients with both PV and ET and is usually recommended for intermediate-risk patients with ET. Situations that should prompt consideration of cytoreductive therapy in low-risk patients with PV and very-low-/low-risk patients with ET are detailed above. The platelet count reaching ⩾1500 × 109/l is an additional indication for cytoreductive therapy in ET with a goal to reduce bleeding, not clotting risk. 6 Worldwide, hydroxyurea (HU) is the most widely used cytoreductive agent for patients with both PV and ET, based on the findings of the PT-1 randomized controlled trial (RCT) in 809 high-risk patients with ET, in which HU was superior to anagrelide in terms of rates of arterial thrombosis, serious hemorrhage and myelofibrotic progression. 36 Low-dose aspirin is usually recommended, 37 unless there is evidence of aVWD. Anagrelide was non-inferior to HU in the phase III ANAHYDRET trial (n = 259), but aspirin use was not mandated in this trial and diagnosis of ET was per WHO 2008 criteria, important differences with the PT-1 trial. 38 Although never proven, persistent doubts over the long-term leukemogenicity of HU has led experts to recommend consideration of interferon as an alternative in young patients with long life expectancy.6,27 Recombinant interferon alfa is also a reasonable therapeutic choice after HU failure in both PV and ET, as are ruxolitinib in PV and anagrelide in ET.6,27 A large, open-label RCT (n = 382) recently found no benefit to the addition of HU to aspirin in patients with ET 40–59 years of age who lacked any high-risk features, that is, a history of ischemia, thrombosis, embolism, hemorrhage or extreme thrombocytosis (platelets ⩾1500 × 109/l). 39
Interferons
Interferon alfa has been used successfully for the treatment of PV and ET for many years, with consistently high hematologic as well as molecular remission rates.40,41 Interferon alfa is able to clear not just JAK2- but also CALR-mutated clones, 42 suggesting its potential as a true disease-modifying agent, although there is preclinical evidence that CALR-mutated ET may be less responsive to interferon alfa than JAK2-mutated disease. 43 In general, response rates to interferon are lower in the presence of other, nondriver mutations, and these patients are more likely to exhibit clonal evolution during therapy. 44 Although pegylation offers the convenience of once weekly administration and superior tolerability, discontinuation rates remain high, primarily driven by the unique toxicities of interferon therapy, including flu-like symptoms, depression, hepatotoxicity and autoimmune syndromes. 45 As alluded to above, pegylated interferon alfa represents a reasonable alternative to HU for frontline therapy of both PV and ET in patients needing cytoreduction. Results of the final analysis of the Myeloproliferative Disorders Research Consortium (MPD-RC) 112 global phase III trial of pegylated interferon alfa-2a (n = 82) versus HU (n = 86) in previously untreated patients (HU for <3 months permitted) with high-risk PV or ET (defined according to the 2008 WHO criteria, disease duration <5 years) were recently presented. 46 There were no significant differences between the treatment arms in terms of complete or overall response rates (ORRs) at 12 or 24 months. Interestingly, achievement of a complete hematologic response (CHR) was associated with worsening of some symptom and quality of life parameters. 47 The toxicity profiles of the two agents were different, but toxicity was not a major reason for discontinuation in either arm. Bone marrow pathologic responses were more frequently observed in patients with ET, but there was no significant difference between the two treatment arms. 46 The phase III DALIAH trial compared recombinant interferon alfa (-2a or -2b, n = 35) to HU (n = 21) in PV patients over 60 years of age. 48 The CHR rate at 36 months was significantly higher in the interferon group (49% versus 19%, p = 0.045), but the rates of hematocrit control and molecular response (all partial) were not. Significantly higher proportions of patients in the interferon group maintained CHR (40% versus 7%, p = 0.048) and partial molecular response (91% versus 29%, p = 0.01) at 36 months. Treatment discontinuation was significantly more frequent among interferon-treated patients (p = 0.03). 48 The MPD-RC has also studied pegylated interferon alfa-2a in patients with high-risk PV (n = 50) or ET (n = 65) in the second-line setting after failure (resistance or intolerance) of HU. 49 The ORR at 12 months was 69.2% in patients with ET and 60% in those with PV. The best ORR by intention-to-treat analysis was 70.8% for ET and 64% for PV. Overall, eight patients (11.1%) achieved a bone marrow complete response and seven of these eight patients had a hematologic response. The rate of CHR was higher among CALR-mutated patients (56.3% versus 28.6%, p = 0.02). 49
Ropeginterferon alfa-2b (peg-proline-interferon alfa-2b) is a novel, monopegylated interferon that can be administered once every 2 weeks. In the single-arm, open-label phase I/II PEGINVERA study in 51 patients with PV, this agent produced an ORR of 90%, with 47% complete and 43% partial responses. 50 The rates of best molecular response were complete (CMR) in 21% and partial in 47%. Overall, three patients, two of whom had more than one chromosomal aberration, achieved complete cytogenetic responses. 51 This agent was then taken forward into a phase III registrational trial in Europe, termed the PROUD/CONTI-PV trial. Aiming to recruit an ‘early’ PV population, the investigators enrolled both treatment-naïve high-risk patients requiring cytoreduction as well as those who had received HU for <3 years but had not achieved a CHR. The initial part of the trial (PROUD-PV) was designed to show non-inferiority of ropeginterferon alfa-2b to HU in terms of CHR rate at 12 months, and met its primary endpoint. 52 In the second part of the trial (CONTI-PV), 95 patients received ropeginterferon alfa-2b and 76 received best available therapy (BAT). 53 Cytopenias were more frequent in the control group, while increased liver enzymes and myalgias were more prominent in the interferon group. CHR rates were numerically higher after 12 months of treatment in the HU group (75% versus 62.1%, p = 0.12), while at 2 and 3 years, CHR rates significantly favored ropeginterferon alfa-2b (70.5%) over BAT (≈50%, p = 0.01). At 3 years, the rate of CHR plus improvement in disease burden (splenomegaly and symptoms) was also significantly higher for ropeginterferon alfa-2b than for BAT (52.6% versus 37.8%, p = 0.04), while it was nonsignificantly higher in the HU arm after 12 months (51.3% versus 46.3%, p = 0.5), underscoring the fact that responses to interferon take time. A very similar pattern was observed with regard to reduction of the JAK2 V617F allele burden, with numerically more patients in the HU arm having a molecular response by 12 months (50.7% versus 43.6%, p = 0.5), while 66% of ropeginterferon alfa-2b patients compared with 27% of BAT patients had achieved a molecular response by 3 years (p < 0.0001). In contrast with previously reported findings with interferon alfa-2a, 54 significant declines in non-JAK2 mutant allele burdens were observed on ropeginterferon alfa-2b. 53 Ropeginterferon alfa-2b (Besremi) has since received a ‘positive opinion’ from the European Medicines Agency, recommending the granting of marketing authorization for the drug for the treatment of PV without symptomatic splenomegaly. 55
Ruxolitinib
The JAK1/2 inhibitor ruxolitinib is currently approved for the treatment of patients with PV whose disease is resistant to or who are intolerant of HU, based on superiority over BAT demonstrated in the RESPONSE and RESPONSE-2 studies in patients with and without splenomegaly, respectively.56,57 Although the formal European LeukemiaNet (ELN) definition of HU resistance requires a dose of 2 g daily for at least 3 months, 58 real-world studies indicate that this dose is infrequently achieved. 59 Resistance to HU as defined by the ELN, and in particular, having no response in leukocyte count, has been associated with a higher risk of death and leukemic transformation. 60 Among the different criteria for HU intolerance, the development of cytopenias at the lowest dose of HU needed to achieve a complete or partial response appears to be associated with a higher risk of myelofibrotic progression, leukemic transformation and death. 61 While the need for phlebotomies on HU would intuitively suggest poorly controlled/more proliferative disease, studies have arrived at opposite conclusions as to whether this has a deleterious impact on the incidence of thrombotic events.62,63 In RESPONSE, the rates of hematocrit control, spleen volume reduction (SVR), CHR and ⩾50% reduction in total symptom score (TSS) at week 32 were 60% and 20%, 38% and 1%, 24% and 9%, and 49% and 5%, for ruxolitinib and BAT, respectively. 56 There was a trend towards fewer thrombotic events in the ruxolitinib group. An interesting biologic correlate of this may be the inhibition by JAK inhibition of neutrophil extracellular trap formation, which is implicated in thrombogenesis in the MPNs. 64 There was also a progressive decline in the JAK2 V617F allele burden in both ruxolitinib-randomized and crossover patients, although the clinical significance of this remains unclear. 65 Crossover was permitted after week 32, the time point at which the primary endpoint, a composite of hematocrit control and spleen volume reduction (SVR), was assessed, and no patient remained on BAT after week 80. The 5-year follow-up data from the RESPONSE trial were recently presented. 66 At the final analysis, the Kaplan–Meier estimated probability of maintaining the primary response for 224 weeks (starting from week 32) in the ruxolitinib arm was 0.74, and that of maintaining clinicohematologic response (hematocrit control without phlebotomy, platelets ⩽ 400 × 109/l, leukocytes ⩽ 10 × 109/l, SVR ⩾ 35%) was 0.67, and the median durations of the primary and clinicohematologic responses had not been reached. No new safety signals emerged. The primary endpoint in RESPONSE-2 was hematocrit control at week 28, achieved by 62% of patients in the ruxolitinib arm compared with 19% of patients in the BAT arm (p < 0.0001). 57 Crossover was permitted after week 28, and no patient remained on BAT by week 80. Among hematocrit responders at week 28, the probability of maintaining the response at week 80 was 78%, and durable CHR was achieved in 24% of patients in the ruxolitinib arm versus 3% in the BAT arm at week 80. 67 Ruxolitinib efficacy and safety, both after and versus interferon use in the RESPONSE and RESPONSE-2 studies, was examined in an ad hoc analysis, and ruxolitinib was found superior in terms of both efficacy and tolerability when compared with interferon as BAT; furthermore, ruxolitinib was efficacious in patients who received interferon as BAT but did not respond and crossed over to receive ruxolitinib. 68 Patients with PV who have splenomegaly, are on HU or require phlebotomy have high symptom burdens, 69 and blood count control does not necessarily correlate with symptom control. 70 The RELIEF study compared (1:1) continuation of HU with switching to ruxolitinib in 110 symptomatic patients with PV whose blood counts were well controlled on a stable dose of HU. 71 The primary endpoint, a ⩾50% reduction in the TSS cytokine symptom cluster (tiredness, itching, muscle aches, night sweats and sweats while awake) at week 16, was achieved by 43.4% of patients in the ruxolitinib arm and 29.6% of patients in the HU continuation arm (p = 0.139). Reactivation of varicella zoster virus (shingles) and nonmelanoma skin cancers, particularly in patients with a history of the same, are important adverse events (AEs) that call for vigilance when treating patients with PV with ruxolitinib, and shingles vaccination using the inactivated (killed) virus should be considered in all patients.
Ruxolitinib is not currently approved for ET, although several trials are ongoing (ClinicalTrials.gov identifiers: NCT02577926, NCT02962388, NCT03123588). Ruxolitinib was compared with BAT in 110 patients with ET and HU resistance or intolerance in the investigator-initiated MAJIC-ET RCT in the United Kingdom. 72 At 1 year, there was no significant difference in CHR rates (46.6% with ruxolitinib and 44.2% with BAT, p = 0.4), and rates of thrombosis, hemorrhage and transformation were no different at 2 years. However, improvement in disease-related symptoms was greater in patients receiving ruxolitinib. Ruxolitinib was also studied in 39 HU-resistant/intolerant patients with ET in an open-label, single-arm trial. 73 Rapid decreases in leukocyte and platelet counts through the first 4 weeks of therapy were noted, followed by normalization/stabilization, and ⩾50% improvements in ET-related symptom scores, for example, bone pain, pruritus, night sweats, numbness/tingling and weakness between baseline and after 12 weeks of therapy occurred in the majority of patients. The recently published results of the MPN Landmark Survey have drawn attention to the symptom burden faced by patients with ET, PV and MF, and their relative underappreciation by physicians.74–76
Investigational approaches: human double minute 2 inhibition and other strategies
Human double minute 2 (HDM2) is the physiologic negative regulator of p53 and small-molecule inhibition of HDM2 is being pursued as a therapeutic strategy in cancer types where deletion or mutation of TP53 is uncommon, such as de novo AML, as a means of restoring the function of wild-type TP53 and inducing apoptosis. Although TP53 mutations are common in the blast phase of MPNs, 77 they are quite rare in chronic-phase disease, especially in PV and ET. Furthermore, JAK2 V617F enhances the expression of HDM2 in MPNs. 78 Idasanutlin is an orally bioavailable antagonist of HDM2 that preferentially eliminated JAK2 V617F+ MPN hematopoietic progenitor cells, both alone and synergistically with pegylated interferon alfa-2a, and pretreatment of PV and PMF CD34+ cells decreased donor-derived chimerism as well as JAK2 V617F allele burden upon transplantation into immunodeficient mice in preclinical studies. 79 These findings were subsequently translated into an investigator-initiated phase I trial in patients with high-risk, JAK2-mutated PV (n = 11) or ET (n = 1) who were resistant/intolerant to at least one prior therapy (HU, interferon or anagrelide). 80 Baseline HDM2 levels were higher in study participants than in healthy controls, and plasma MIC-1 levels were significantly increased in the six patients tested following treatment with idasanutlin, signaling activation of the p53 pathway. Idasanutlin was given on days 1–5 of each cycle, and pegylated interferon alfa-2a could be added after six cycles of idasanutlin monotherapy in patients with a suboptimal response. No grade 4 nonhematologic AE occurred at either the 100 mg or 150 mg daily dose of idasanutlin, and no hematologic AE of any grade occurred. Grade 3 headache, fatigue and pain occurred in one patient each, but no grade 3/4 gastrointestinal AE was observed. Anti-emetic prophylaxis was routine. The ORR by ELN-International Working Group (IWG) criteria 81 was 75% (9 of 12), with 7 responders (4 complete and 3 partial) in part A (idasanutlin monotherapy) and 2 additional responders (1 complete and 1 partial) in part B (idasanutlin plus pegylated interferon alfa-2a). The TSS declined by ⩾50% in most patients, and the median reduction in the mutant JAK2 allele burden was 43%. Bone marrow histomorphologic responses were also observed. A patient with a p53 mutation at baseline did not respond. Idasanutlin is now being tested in a global, single-arm phase II trial in patients with HU-resistant/intolerant PV (ClinicalTrials.gov identifier: NCT03287245). A more potent oral agent, KRT-232, is also being explored in a two-part phase II study in phlebotomy-dependent patients with PV (ClinicalTrials.gov identifier: NCT03669965). Part A of this trial is a dose- and schedule-finding portion in HU-resistant/intolerant or interferon-pretreated patients, while part B is planned to be a randomized comparison of KRT-232 (at the optimal dose and schedule to be derived from part A) to ruxolitinib in HU-resistant/intolerant patients with splenomegaly, as in RESPONSE.
Histone deacetylase inhibitors (HDACis), for example, vorinostat, givinostat, are active in PV, both in murine models 82 and in the clinic,83,84 but the chronic, low-grade toxicities of these agents make them difficult to administer for long periods, making the future of this class of agents uncertain in MPNs as a whole. 85 The telomerase inhibitor imetelstat produced high rates of best hematologic response (100%; 89% CHR) and molecular response in a small study in patients with previously treated ET (n = 18), 86 but toxicity was significant, and current development of this agent in MPNs is focused on MF. 87 The same is true of the recently resurrected JAK1/2 inhibitor momelotinib which, while promising in terms of anemia and symptom benefits in patients with MF,88,89 had limited efficacy in patients with PV or ET. 90
Conclusion
After many years of a relative lack of progress in PV and ET, the regulatory approval of ruxolitinib and the impending approval (in the European Union) of ropeginterferon alfa-2b for PV represent important therapeutic advances that have kindled interest in investigating other, mechanism-based agents, for example, HDM2 inhibitors, for the treatment of these largely indolent conditions. From a clinical practice standpoint, increasing awareness of the deleterious effects of leukocytosis and the symptom burden faced by patients with PV and ET may influence practice patterns and drug development efforts. Ultimately, patients and providers alike await the arrival of agents that can modify the underlying disease biology so as to prevent progression to MF and transformation to AML.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
PB reports honoraria from Incyte Corporation, Celgene Corporation and Blueprint Medicines Corporation, and research funding from Incyte Corporation, Celgene Corporation, CTI BioPharma, Kartos Therapeutics, Constellation Pharmaceuticals, Pfizer, Inc., Astellas Pharmaceuticals, NS-Pharma, Promedior and Blueprint Medicines Corporation.