
Abstract
Mutations in the fms-like tyrosine kinase 3 (FLT3) gene are detected in approximately one-third of patients with newly diagnosed acute myeloid leukemia (AML). These consist of the more common FLT3-internal tandem duplication (ITD) in approximately 20–25% of AML cases, and point mutations in the tyrosine kinase domain (TKD) in approximately 5–10%. FLT3 mutations, especially FLT3-ITD, are associated with proliferative disease, increased risk of relapse, and inferior overall survival when treated with conventional regimens. However, the recent development of well tolerated and active FLT3 inhibitors has significantly improved the outcomes of this aggressive subtype of AML. The multikinase inhibitor midostaurin was approved by the United States Food and Drug Administration (US FDA) in April 2017 for the frontline treatment of patients with FLT3-mutated (either ITD or TKD) AML in combination with induction chemotherapy, representing the first new drug approval in AML in nearly two decades. In November 2018, the US FDA also approved the second-generation FLT3 inhibitor gilteritinib as a single agent for patients with relapsed or refractory FLT3-mutated AML. Promising phase I and II efficacy data for quizartinib is likely to lead to a third regulatory approval in relapsed/refractory AML in the near future. However, despite the significant progress made in managing FLT3-mutated AML, many questions remain regarding the best approach to integrate these inhibitors into combination regimens, and also the optimal sequencing of different FLT3 inhibitors in various clinical settings. This review comprehensively examines the FLT3 inhibitors currently in clinical development, with an emphasis on their spectra of activity against different FLT3 mutations and other kinases, clinical safety and efficacy data, and their current and future roles in the management of AML. The mechanisms of resistance to FLT3 inhibitors and potential combination strategies to overcome such resistance pathways are also discussed.
Keywords
Introduction
Acute myeloid leukemia (AML) is a genomically heterogeneous disease characterized by a number of well-described recurrent mutations that drive disease phenotype, response to therapy, and risk for relapse. 1 Mutations of the fms-like tyrosine kinase 3 (FLT3) gene are the most common genomic alterations in AML, identified in approximately one-third of newly diagnosed patients.2–4 FLT3-internal tandem duplication (ITD)-mutated AML often presents with proliferative features such as leukocytosis and increased peripheral blasts. When treated with combination chemotherapy alone, FLT3-ITD mutated AML is associated with a higher rate of relapse and inferior overall survival than FLT3 wildtype disease.5–11 In contrast the prognostic impact of FLT3 tyrosine kinase domain (TKD) mutations is less clear. This has led to intense interest in the development of targeted agents against the FLT3 mutant protein, eventually culminating in both United States Food and Drug Administration (US FDA) and European Medicines Agency (EMA) approvals of the multikinase tyrosine kinase inhibitor (TKI) midostaurin in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation in adults with newly diagnosed FLT3-mutated AML. This was the first AML drug to receive regulatory approval in the US since 2000. Following the midostaurin approval, in November 2018, the second-generation FLT3 inhibitor gilteritinib was approved by the US FDA for use as a single agent for adults with relapsed or refractory FLT3-mutated AML. Several other FLT3 inhibitors are currently in advanced clinical development with anticipated approval in different AML settings and are likely to change the therapeutic approach to this subtype of AML in the very near future.
In this review, we will discuss the landscape of FLT3 mutations in AML and how these mutations impact prognosis. We will also review the available clinical data on the several FLT3 inhibitors currently in development and describe the potential role that each of these FLT3 TKIs may place in the future management of FLT3-mutated AML, with particular emphasis on their distinct mechanism and spectra of action. Finally, we will discuss the ongoing studies of these various inhibitors and how rationally designed combination therapies may overcome the known mechanisms of resistance to FLT3 inhibitors in AML.
Wildtype and mutated FLT3 in AML
The FLT3 protein is a receptor tyrosine kinase widely expressed in hematopoietic progenitor cells. Upon binding to the cytokine FLT3 ligand, FLT3 receptors dimerize; this dimerization leads to conformational changes in the FLT3 proteins that expose their adenosine triphosphate (ATP) pockets and trigger autophosphorylation and signal transduction. Downstream effects of this intracellular signaling include the promotion of cellular proliferation and survival and inhibition of differentiation.12,13 Independent of the presence or absence of FLT3 mutations, FLT3 is commonly overexpressed in lineage-restricted AML blasts and may be associated with worse outcomes, suggesting that it may serve as a therapeutic target irrespective of FLT3 mutation status.14,15
FLT3 mutations (ITD, TKD, or both) are identified in approximately one-third of patients with newly diagnosed AML.2–4 These mutations are particularly enriched in patients with a normal karyotype.8,16 However, as they are a later event in leukemogenesis and generally not a primary, initiating event, FLT3 mutations are not restricted to any particular AML subgroups. 17 ITD mutations are identified in 20–25% of patients with newly diagnosed AML, whereas point mutations in the TKD are identified in 5–10%, approximately half of which occur at D835 in the activation loop.18,19 Regardless of the type of mutation, both cause spontaneous dimerization and ligand-dependent growth. Importantly, the mutated receptor remains responsive to the FLT3 ligand, which is capable of further modulating signaling from the mutant kinase. 20
Prognostic impact of FLT3 mutations
Prior to the advent of FLT3 inhibitors, studies have consistently shown that FLT3-ITD-mutated AML is associated with an increased risk of relapse and worse survival compared with wildtype disease.5–11 In contrast, the clinical relevance of TKD mutations is less clear,21–24 although it is generally accepted that their prognostic impact is neutral and that the presence of a TKD mutation should not alter risk assessment. 25 Among patients with FLT3-ITD mutations, co-mutations further influence outcomes, particularly the presence of a co-existing NPM1 mutation, which is associated with a decreased risk of relapse and improved survival in FLT3-ITD-mutated AML, especially in patients who have a low allele burden FLT3-ITD.2,4,26–29 The allelic ratio of FLT3-ITD to wildtype FLT3 has been shown to strongly influences outcomes in several studies of chemotherapy-based induction therapy (3+7 or similar) in patients with newly diagnosed FLT3-ITD-mutated AML.29–31 The FLT3-ITD allelic ratio is generally defined as the ratio of the area under the curve of ‘FLT3-ITD’ divided by the area under the curve of ‘FLT3-wildtype’ using a semi-quantitative DNA fragment analysis. 25 A higher FLT3-ITD ratio (generally defined as ⩾0.5 or ⩾0.7, depending on the study) is generally associated with worse survival than lower ratios, likely reflecting increased FLT3 dependency in the cases with high allelic ratios. It is important to note that most of these studies on the impact of FLT3-ITD allelic ratio did not incorporate an FLT3 inhibitor as part of the induction, consolidation, or maintenance therapy; however, one study that included midostaurin in the treatment regimen failed to show a difference in the cumulative incidence of relapse according to FLT3-ITD allelic ratio of <0.5 versus ⩾0.5. 32 Given consistent evidence of the cooperative role of NPM1 mutations and the FLT3-ITD allelic ratio in influencing outcomes in AML across most studies, both have been incorporated into consensus recommendations on genomic-based risk stratification. 25 It has been suggested that a longer ITD length may be associated with a higher risk of relapse, although studies have yielding conflicting results16,33,34 and therefore ITD length assessment is not recommended for routine risk stratification.
The primary importance of accurate risk stratification in AML is to determine appropriate post-remission strategies, particularly to decide between consolidative chemotherapy or allogeneic hematopoietic stem cell transplantation (HSCT) in first remission. 1 With the exception of one study that was limited by small numbers of FLT3-mutated patients who underwent HSCT and an unexpectedly high transplant-related mortality, 35 the majority of evidence suggests that HSCT improves outcomes for patients with FLT3-ITD-mutated AML, when patients were analyzed agnostic of NPM1 status and allelic ratio.36–40 Furthermore, the RATIFY study showed that the addition of midostaurin to chemotherapy proportionally improved overall survival in FLT3 allele low (<0.5) and FLT3 allele high (⩾0.5) patients, albeit the improvement in these subgroups was not statistically significant (p = 0.19 for both analyses). 41 Therefore, at most centers, fit patients with FLT3-ITD mutations are generally considered for HSCT in first remission, with the possible exception of NPM1-mutated patients with a low allelic burden FLT3-ITD mutation, as this constitutes a relatively favorable risk group in the pre-TKI era. 25 In the absence of robust prognostic analysis in patients treated with FLT3 inhibitors in induction/consolidation, the current standard at our institution and at many academic centers in the US has been to add a multikinase FLT3 inhibitor (midostaurin or sorafenib) to induction and consolidation in patients with FLT3-mutated AML (defined as FLT3 mutation on next-generation sequencing or FLT3 allele burden >3% on polymerase chain reaction) and to proceed to HSCT in first remission for patients with FLT3-ITD-mutated AML that is classified as intermediate or adverse risk by European LeukemiaNet guidelines. 25 However, studies reassessing the role of HSCT in patients with FLT3-mutated AML who receive frontline FLT3 inhibitors are needed. Randomized clinical trial data supports the addition of midostaurin, 41 which is US FDA approved for this indication, while we generally add off-label sorafenib (unless a TKD mutation is present) based on data from phase II studies showing its safety and efficacy in combination with chemotherapy.42–44 We also have routinely used post-HSCT maintenance with a FLT3 inhibitor, either sorafenib (most commonly used as maintenance in our center) or crenolanib (in the setting of a previous clinical trial with post-HSCT crenolanib). Recent randomized data from ASH 2018 suggest that post-HSCT sorafenib maintenance significantly improved overall survival (OS) in FLT3-ITD-mutated patients who underwent HSCT. 45
Characteristics of FLT3 inhibitors and principles of use
Given the established pathobiological and prognostic role that FLT3-ITD mutations play in AML, mutant FLT3 is an attractive therapeutic target for leukemia-directed therapies. 46 To this end, several FLT3 inhibitors have been developed and are currently in clinical trials (Table 1). These agents work largely through competitive inhibition of ATP-binding sites in the FLT3 receptor, leading to cell cycle arrest and differentiation. 47 In addition, FLT3 TKIs vary in their ability to target non-FLT3 signaling pathways, which influences both the tolerability and the efficacy of different agents. For example, first-generation FLT3 inhibitors (e.g. lestaurtinib, sorafenib, and midostaurin) are less specific for FLT3 and have broad kinome profiles with more off-target toxicities; in contrast, second-generation FLT3 inhibitors (e.g. quizartinib, crenolanib, and gilteritinib) are more specific and potent at inhibiting FLT3 with narrower kinome profiles.48,49
Characteristics of FLT3 inhibitors currently in clinical development.
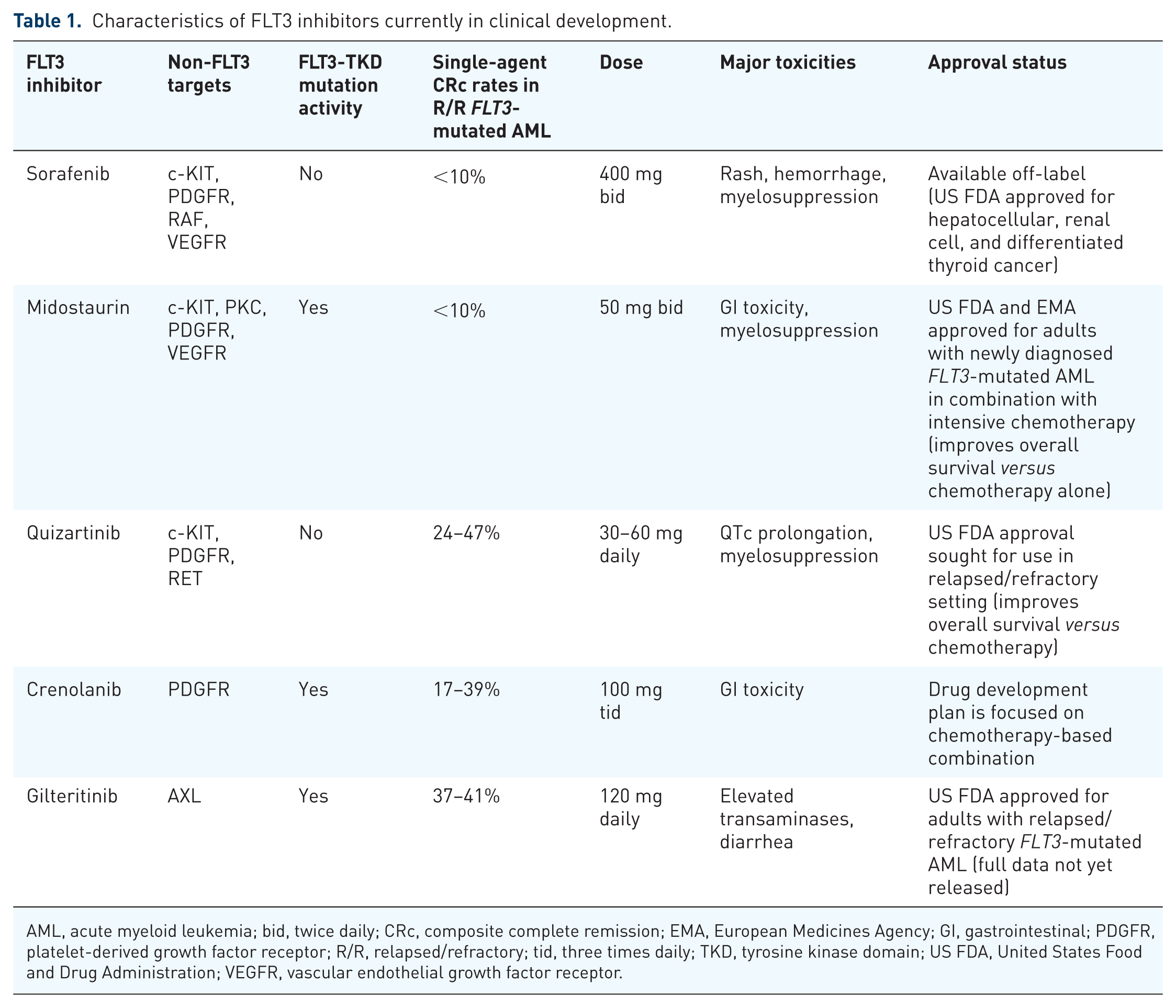
AML, acute myeloid leukemia; bid, twice daily; CRc, composite complete remission; EMA, European Medicines Agency; GI, gastrointestinal; PDGFR, platelet-derived growth factor receptor; R/R, relapsed/refractory; tid, three times daily; TKD, tyrosine kinase domain; US FDA, United States Food and Drug Administration; VEGFR, vascular endothelial growth factor receptor.
In addition to their off-target effects, FLT3 TKIs have varying potencies against different FLT3 mutations. 50 Type I inhibitors (e.g. lestaurtinib, midostaurin, gilteritinib, and crenolanib) bind to the gatekeeper domain of FLT3 near the activation loop or the ATP-binding pocket regardless of receptor conformation, while type II inhibitors (e.g. sorafenib and quizartinib) bind to the hydrophobic region directly adjacent to the ATP-binding domain when the protein is in its inactive conformation. Notably, type II FLT3 inhibitors do not have significant activity against TKD mutations, as these mutations favor the active protein conformation. 51 As FLT3-TKD mutations may be present at the time of diagnosis or emerge as a mechanism of resistance under the therapeutic pressure of chemotherapy or FLT3 inhibitor therapy, the inhibitory activity of these agents against TKD mutations should be a consideration when selecting the optimal FLT3 inhibitor for a particular patient, especially in the setting of an established TKD mutation.
While several FLT3 inhibitors have single-agent activity, there is preclinical rationale supporting the combination of these agents with cytotoxic chemotherapy, particularly given concomitantly or immediately after chemotherapy. 52 Several studies have evaluated such combinations, most notably the randomized phase III RATIFY study of standard chemotherapy with or without midostaurin for patients with newly diagnosed FLT3-mutated AML, which showed an OS benefit with the addition of midostaurin to standard therapy. 41
The efficacy of FLT3 inhibition in AML may also be influenced by the properties of the specific TKI and whether the leukemia is previously untreated or relapsed/refractory. Previously untreated FLT3-mutated AML is often a polyclonal disease, of which the FLT3 mutation may be present only in one subclone, whereas relapsed/refractory AML is a more monoclonal disease. 53 In the former setting in which aberrant FLT3 signaling may be only one of several factors driving the disease biology and phenotype, it has been hypothesized that agents capable of inhibiting multiple kinases in addition to FLT3 (e.g. sorafenib and midostaurin) may be particularly beneficial. In contrast, in relapsed/refractory FLT3-mutated AML, there is often a higher FLT3 allelic burden and increased addiction to aberrant FLT3 signaling.54,55 In this setting, the use of more selective and potent second-generation FLT3 inhibitors may be ideal. However, the clinical data at this time do not clearly indicate whether a broad or selective kinase FLT3 inhibitor would be better during induction and relapse, respectively. A number of clinical trials combining selective FLT3 inhibitors (e.g. quizartinib, gilteritinib, and crenolanib) with induction therapy in patients with newly diagnosed FLT3-mutated AML are ongoing and will likely help answer this question (ClinicalTrials.gov identifiers: NCT02668653, NCT02752035, NCT02283177, NCT03258931)
Clinical experience with FLT3 inhibitors
Lestaurtinib
Lestaurtinib is a first-generation FLT3 inhibitor and one of the first to be studied in clinical trials. It has activity against FLT3, as well as JAK2 and Trk tyrosine kinases. In early studies of single-agent lestaurtinib, including a phase I/II study in relapsed/refractory AML (n = 17) and a phase II study of older adults with newly diagnosed AML (n = 29, including 5 patients with an activating FLT3 mutation), transient reductions of bone marrow blasts were observed. No patients achieved complete remission (CR) or complete remission with incomplete count recovery (CRi).56,57 These studies were followed by two randomized phase III trials evaluating chemotherapy with or without the addition of lestaurtinib in patients with FLT3-mutated AML, including one study in patients in first relapse 58 and one in younger patients with newly diagnosed AML. 59 Neither of these trials showed an improvement in either response rates or survival with the addition of lestaurtinib to chemotherapy in the intention to treat population, which may have in part been due to suboptimal FLT3 inhibition. Among patients in the frontline lestaurtinib plus chemotherapy trial in whom >85% FLT3 inhibition was sustained with lestaurtinib therapy, a benefit in relapse rate and OS was observed, supporting the potential for FLT3 inhibitor-based regimens in this context. 59 However, given these negative phase III study results, lestaurtinib is no longer in clinical development.
Sorafenib
Sorafenib is another first-generation FLT3 inhibitor that has a wide variety of kinase inhibitory activity, including c-KIT, platelet-derived growth factor receptor (PDGFR), and vascular endothelial growth factor receptor (VEGFR). As a type II inhibitor, it notably has no clinically significant TKD activity, which limits its utility in patients harboring FLT3-D835 and other FLT3 point mutations. Like lestaurtinib, sorafenib had limited single-agent activity in AML, with marrow remissions observed in <10% of patients treated with sorafenib monotherapy.60–62
The clinical results with sorafenib have been more promising when used in combination, both with cytotoxic chemotherapy and with the hypomethylating agent azacitidine. In a phase II study of 62 adults with newly diagnosed AML (median age 53 years; range, 18–66 years), the combination of sorafenib with idarubicin and cytarabine (IA regimen) resulted in an overall CR or CR with incomplete platelet recovery (CRp) rate of 87%.42,43 A total of 23 patients (37%) harbored a FLT3-ITD mutation, and the CR/CRp rates for those with and without mutation were 95% and 84%, respectively. There was no difference in survival outcomes between patients with and without FLT3-ITD mutation, suggesting that the addition of sorafenib benefitted these patients by improving the survival in traditionally adverse risk FLT3-ITD patients to that of non-FLT3-ITD-mutated patients. The phase II CALGB 11001 study evaluated standard cytarabine and anthracycline-based induction and consolidation chemotherapy in combination with sorafenib in 54 patients age ⩾60 with newly diagnosed FLT3-mutated AML. Among patients with FLT3-ITD mutations (71% of the cohort), the median OS was 15.0 months and the 1-year OS rate was 62%, which was significantly better than the historical control group (1-year OS rate 30%, p < 0.0001). Together, these studies suggest that the combination of sorafenib with intensive chemotherapy is safe and may improve outcomes in both younger and older adults with FLT3-ITD-mutated AML.
The combination of sorafenib with azacitidine has also been shown to be well tolerated and effective. In a study of this combination in 43 adults with predominantly relapsed/refractory FLT3-ITD-mutated AML, the overall response rate was 46%. 63 Notably, among the 40 patients with FLT3-ITD mutations, 9 had prior treatment with at least one FLT3 TKI. Despite the encouraging response rate, the median duration of response was only 2.3 months and the median OS was 6.2 months. Additional translational work showed that FLT3 ligand levels did not rise appreciably with exposure to the combination regimen. This finding is important as increased FLT3 ligand has been observed in patients treated with chemotherapy and has been suggested to be a mechanism of FLT3 inhibitor resistance in these patients. 20 Despite the short duration of response and survival, these correlative studies suggest that hypomethylating agent and FLT3 inhibitor-based combinations may potentially overcome this mechanism of resistance, although the clinical significance remains to be determined. In a similar study in the frontline setting in 27 older adults with FLT3-ITD-mutated AML who were not candidates for intensive induction therapy (median age 74 years; range, 61–86 years), azacitidine plus sorafenib resulted in an overall response rate of 78% with a median duration of response of 14.5 months and median OS of 8.3 months. 64
Given its multikinase activity, sorafenib has also been evaluated in combination with cytotoxic chemotherapy in patients with AML irrespective of FLT3 status. The SORAML study randomized 267 patients ⩽60 years of age with newly diagnosed AML to standard intensive induction and consolidation chemotherapy or to the same chemotherapy regimen in combination with sorafenib followed by 1 year of sorafenib maintenance. 44 Only 17% of the randomized patients had a FLT3-ITD mutation at diagnosis. Patients randomized to the sorafenib-containing regimen had an improvement in both event-free survival (EFS) and relapse-free survival (RFS) compared with the placebo arm (3-year EFS rate: 22% versus 40%, p = 0.01; 3-year RFS rate: 56% versus 38%, p = 0.017), although no OS benefit was observed. The addition of sorafenib was associated with increased adverse events, particularly diarrhea, bleeding, cardiac events, hand-foot-skin reaction and rash but the 30-day mortality was comparable between the two arms (2% in the sorafenib arm and 1% in the placebo arm). In an update with longer follow up (median: 78 months), EFS and RFS benefits with the addition of sorafenib continued to be observed. 65 Additionally, there appeared to be a late separation in the OS curves after 2 years, with a 5-year OS rate of 61% and 52% (p = 0.28) for the sorafenib and placebo arms, respectively. Overall, the results from the SORAML study suggest that there may be a benefit to adding a multikinase inhibitor such as sorafenib to induction even among patients without FLT3-ITD mutations. In contrast, a similar randomized study in an older population of patients with newly diagnosed AML (median age 68 years; range, 61–80 years) showed no EFS or OS benefit in this elderly population, likely driven by increased early deaths in the sorafenib arm (17% versus 7% with the placebo arm). 66 Thus, the benefit of adding sorafenib to chemotherapy regardless of FLT3 status in frontline treatment regimens may be most prominent in younger, fitter patients who likely have a higher tolerance and resilience to addition of therapies to induction.
Midostaurin
Midostaurin is a first-generation FLT3 inhibitor capable of targeting ITD and TKD mutations with broad-spectrum inhibitory activity against VEGFR, protein kinase C, c-KIT, and PDGFR-β. 67 In a study of 95 patients, midostaurin single-agent therapy in relapsed/refractory AML resulted in blast reductions in 71% of patients with FLT3-mutant AML and 42% of patients with FLT3-wildtype, although no CR or CRis were observed. 68 In a smaller study of 20 patients with relapsed/refractory FLT3-mutated AML, 2 patients (10%) achieved bone marrow blast reduction <5%. In a phase Ib study in 69 younger adults with AML, the addition of midostaurin to standard cytarabine and anthracycline induction and consolidation was found to be safe and effective, with a CR rate of 80% (74% for FLT3 wildtype versus 92% for FLT3-mutated). 44 OS was similar regardless of FLT3 status, suggesting the addition of midostaurin overcame the poor prognostic impact of the FLT3 mutation.
The encouraging findings of this dose-finding study led to a large, multinational, randomized phase III RATIFY (CALBG 10603) study. 69 In this study, 717 adults <60 years of age with newly diagnosed FLT3-mutated AML (either ITD or TKD) were randomized to receive standard induction followed by consolidation chemotherapy (or HSCT, if indicated) with either midostaurin or placebo. Midostaurin or placebo were given on Days 8–21 of each induction and consolidation cycle, followed by up to an additional one year of maintenance with midostaurin or placebo. A total of 77% of patients had an ITD mutation and 23% had a TKD mutation. Patients in the midostaurin arm had a significant improvement in OS (4-year OS rate: 51.4% versus 44.3%; median OS: 74.7 months versus 25.6 months; p = 0.009), an effect that was seen regardless of type of FLT3 mutation or the ITD allelic burden (i.e. <0.5 or ⩾0.5).
Based on these results, midostaurin was approved by the US FDA in April 2017 and by the EMA in September 2017 for the treatment of adults with newly diagnosed FLT3-mutated AML in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation. Despite use of up to 1 year of maintenance midostaurin in the RATIFY trial, no US FDA approval was given for midostaurin maintenance therapy, whereas use of midostaurin as maintenance was included in the EMA approval. Notably, along with the approval of midostaurin, the US FDA also approved the LeukoStrat CDx FLT3 Mutation Assay (Invivoscribe, Inc. San Diego, CA) as a companion diagnostic test for the detection of FLT3 mutations.
In the RATIFY trial, the largest survival benefit was observed in patients who underwent subsequent HSCT in first remission, which was performed in approximately 25% of the entire cohort (28.1% in the midostaurin group and 22.7% in the placebo group, p = 0.10). The 4-year OS with midostaurin followed by HSCT in first remission was 63.7%, compared with 55.7% for patients who received placebo followed by HSCT in first remission (p = 0.08), which translated to a 24.3% lower risk of death in the midostaurin group in the context of HSCT in first remission. Although no formal measurable residual disease (MRD) data have been reported as part of the RATIFY study, the finding that midostaurin improves outcomes for patients who received HSCT in first remission suggests that it may result in deeper remissions (i.e. more patients achieving MRD negativity) than placebo, as MRD negativity prior to HSCT has been shown to be a primary predictor of better post-HSCT outcomes in AML. 70
Based on the results from this large, multicenter, randomized trial, rapid assessment and identification of a FLT3-ITD or TKD mutation, addition of midostaurin to induction and consolidation, and HSCT in first remission should be the goals of therapy for most patients. In the absence of strong data to the contrary from future analyses, it is reasonable to defer HSCT in first remission for those patients with low allelic burden FLT3-ITD mutation and concomitant NPM1 mutation, which is a favorable risk group in the pre-FLT3 inhibitor era. However, confirmation of these biomarkers in patients who receive FLT3 (Invivoscribe, Inc, San Diego, CA) inhibitor-based therapies is needed. 29
Quizartinib
Quizartinib is a second-generation TKI that is more selective for FLT3 than the first-generation inhibitors, although it still has activity against other receptor tyrosine kinases such as c-KIT and PDGFR. Its more potent FLT3 inhibition was evident in studies of patients with relapsed/refractory AML treated with single-agent quizartinib, in which composite CR (CRc) rates >40%, including CRs, have been observed.71–73 This activity is in clear distinction to the first-generation FLT3 inhibitors, in which single-agent use in the relapsed/refractory setting resulted in transient blast decreases, generally without CR/CRis. In contrast with midostaurin, crenolanib and gilteritinib, quizartinib does not have significant activity against TKD mutations, which is an established mechanism of resistance in quizartinib-treated patients. In one small study, the development of resistance TKD mutations was observed in eight of eight patients with acquired resistance to quizartinib, although these findings remain to be confirmed in larger trials. 46
In a phase I study of quizartinib in 76 patients with relapsed/refractory AML irrespective of FLT3 status, the overall response rate was 30%, including a CRc rate of 13%. 71 Notably, responses were more frequently observed in FLT3-ITD mutated patients compared with FLT3 wildtype (overall response rate: 53% versus 14%; CRc rate: 24% versus 5%, respectively). The median duration of response was 13.3 weeks. The primary safety concern with quizartinib was QTc prolongation, which was observed in 12% of patients (any grade). Similar response rates were observed in patients with relapsed/refractory AML treated in a subsequent phase II study of quizartinib unselected for FLT3 status. 72 Notably, in this study of 333 patients, a quizartinib dose of 200 mg daily was initially implemented, based on the recommended phase II dose determined in the earlier phase I study. However, due to a higher than acceptable rate of grade 3 QTc prolongation at this dose, the study was later amended to reduce the dose of quizartinib to 135 mg daily for men and 90 mg daily for women.
Given the uncertainty about the optimal dosing of quizartinib, a randomized phase IIb study was undertaken to evaluate two lower doses of quizartinib that had shown efficacy in the phase I trial, in patients with relapsed/refractory FLT3-ITD-mutated AML. 73 A total of 76 patients were randomized to receive quizartinib at a dose of either 30 mg or 60 mg daily, which could be escalated to 60 mg or 90 mg, respectively, due to lack of response or loss of response. The CRc rate in both groups was 47%, and grade 3 QTc prolongation rates were only 3–5% suggesting equivalent CRc rates with significantly lower QTc prolongation than was seen with the higher doses of quizartinib (grade 3 QTc prolongation rate: 10%). Overall, the 60 mg group had a longer duration of remission (9.1 weeks versus 4.2 weeks), higher HSCT rate (42% versus 32%) and longer median OS (27.3 months versus 20.9 months), leading this dose to be selected for future quizartinib studies.
The phase III QuANTUM-R study evaluated single-agent quizartinib versus salvage chemotherapy in AML with FLT3-ITD mutation (ClinicalTrials.gov identifier: NCT02039726). A total of 367 patients were randomized in a 2:1 ratio to receive either single-agent quizartinib (30 mg lead-in, then 60 mg daily after QTc assessment) or investigator’s choice of salvage chemotherapy [this included low-dose cytarabine, MEC (mitoxantrone, etoposide, and cytarabine) or FLAG-Ida (fludarabine, cytarabine, idarubicin, and G-CSF)]. Quizartinib prolonged OS compared with chemotherapy (median OS: 27.0 weeks versus 20.4 weeks; 1-year OS rate: 27% versus 20%; p = 0.0177). These findings represent the first study of an FLT3 inhibitor shown to improve OS in the salvage setting; US FDA approval has been sought by the company that manufactures quizartinib, although a final regulatory decision is still pending.
Quizartinib has also been studied in the frontline and relapsed/refractory settings in combination with chemotherapy or hypomethylating agents. In a phase I/II study of quizartinib with azacitidine or low-dose cytarabine, the combination resulted in an overall response rate of 75% in patients with relapsed/refractory AML harboring a FLT3-ITD mutation; these results appear superior to those observed with quizartinib monotherapy in phase II and III studies. 74 In the same study, 11 of 12 (92%) older patients >60 years of age in the frontline arm achieved response (CRc rate: 83%) with the combination with a median OS of 18.6 months. These results suggest that both response rates and duration of response may be meaningfully improved by combining quizartinib with hypomethylating agents (as was seen with azacitidine and sorafenib) and should be further evaluated in larger studies. A phase I study in 19 patients confirmed the safety and efficacy of quizartinib in combination with intensive chemotherapy in patients with newly diagnosed AML ⩽ 60 years of age. 75 These data served as the basis for the randomized, multinational, phase III QuANTUM-First that is testing intensive chemotherapy with quizartinib or with placebo during induction, consolidation, and up to 1 year of maintenance in patients with FLT3-ITD-mutated AML (ClinicalTrials.gov identifier: NCT02668653).
Crenolanib
Crenolanib is a second-generation FLT3 TKI that is capable of inhibiting both ITD and TKD mutations, with activity against other signaling pathways including PDGFR. In a phase I study in relapsed/refractory FLT3-mutated AML, an overall response rate of 50% (CRc rate: 39%) was achieved among 18 patients with no prior FLT3 inhibitor exposure; among 36 patients who had received prior FLT3 inhibitors, the overall response rate was 31% (CRc rate: 17%). 76 Activity was noted among patients with both ITD and TKD mutations.
The developmental strategy of crenolanib has focused on evaluating this agent in combination with cytotoxic chemotherapy, both in the frontline and the relapse setting. A study of the IA regimen plus crenolanib in 13 patients with relapsed/refractory FLT3-mutated AML resulted in an overall response rate of 36%. 77 In a frontline study of crenolanib in combination with standard ‘7+3’ induction and high-dose cytarabine consolidation in patients with a median age of 55 years (range, 22–74 years), CR/CRi was achieved in 24 of 25 evaluable patients (96%), with 88% of patients achieving CR. 78 In a later analysis of the 29 patients ⩽60 years of age treated on the study, only 2 patients had relapsed (1 systemic, 1 central nervous system only) with a median duration of follow up of 14 months, suggesting durable responses with the combination. A randomized phase III study of chemotherapy with mitoxantrone and cytarabine with or without crenolanib in patients with relapsed/refractory AML with activating FLT3 mutation is currently accruing (ClinicalTrials.gov identifier: NCT02298166). A frontline, randomized, multicenter phase III study evaluating standard 3+7 induction with either crenolanib or midostaurin during induction, consolidation and up to 1 year of maintenance has also been initiated (ClinicalTrials.gov identifier: NCT03258931)
Gilteritinib
Gilteritinib is a dual FLT3/AXL inhibitor that also has clinical activity against TKD mutations, and does not inhibit KIT. 79 Notably, increased Axl-1 expression has been implicated as a resistance mechanism with other FLT3 inhibitors, including midostaurin and quizartinib.80,81 In a phase I/II study of 252 patients with relapsed/refractory AML, including FLT3-wildtype and FLT3-mutated, the overall response rate with gilteritinib was 40% (CRc rate: 30%). 82 An overall response rate of 49% was observed in FLT3-mutated patients (ITD, D835, or both; CRc rate: 37%) compared with 12% in FLT3-wildtype AML. A total of 37% of patients who failed at least one prior FLT3 inhibitor achieved a response, suggesting the potential for gilteritinib to overcome resistance to other TKIs and suggesting that gilteritinib would still be active in patients relapsed/refractory to induction with midostaurin. Among patients with a FLT3 mutation who received ⩾80 mg daily (n = 169), the overall response rate was 52% (CRc rate: 41%), and the median OS was 31 weeks. Gilteritinib was well tolerated, and the maximum tolerated dose was established at 300 mg daily. However, given uniform in vivo target inhibition across all the dose levels studied and a high proportion of patients responding to the 120 mg/day dose, this lower dose was selected for further evaluation. A phase II study of gilteritinib, gilteritinib plus azacitidine, or azacitidine alone for patients with newly diagnosed FLT3-mutated AML who are unfit for intensive chemotherapy is currently accruing patients (ClinicalTrials.gov identifier: NCT02752035). A randomized phase III study of gilteritinib versus salvage chemotherapy in relapsed/refractory FLT3-mutated AML has recently completed accrual (ADMIRAL study; ClinicalTrials.gov identifier: NCT03182244). Based on an interim analysis of this trial that showed a CRc rate of 21% and median time to best response of approximately 3.5 months, the US FDA approved gilteritinib as single-agent therapy for adults with relapsed/refractory FLT3-mutated AML in November 2018; full details of the study findings, including survival data, are eagerly awaited.
Mechanisms of FLT3 inhibitor resistance and potential novel targets
Despite encouraging response rates with FLT3 TKI-based regimens in both the frontline and relapsed/refractory settings, many patients still fail to respond to FLT3 inhibitor therapy or subsequently relapse. To improve these outcomes, an increased understanding of the mechanisms underpinning FLT3 inhibitor resistance is needed. A full description of the numerous mechanisms of FLT3 inhibitor resistance is outside the scope of this manuscript; however, here we outline some of the best described pathways of resistance reported in preclinical and clinical studies.
One such mechanism of resistance is the development of secondary mutations in the FLT3 gene. 83 These mutations are often single amino acid exchanges in the activating loop residues (e.g. D835, I836, D839, Y842) or gatekeeper residues (e.g. F691).84,85 This resistance mechanism is a particular issue for patients treated with the type II inhibitors (e.g. sorafenib or quizartinib) that do not have significant activity against TKD mutations.46,51,86 However, while treatment-emergent TKD mutations are an important mechanism of FLT3 inhibitor resistance, these mutations may not be the major modality of FLT3 inhibitor resistance. For example, in one study, only 22% of patients treated with FLT3 inhibitors developed a new detectable TKD mutation at the time of progression. 87 Other extrinsic alterations also play an important role in the development of FLT3 inhibitor resistance.
Various intracellular and extracellular pathways have been implicated in FLT3 TKI resistance. These serve as rational targets for FLT3 inhibitor-based combination regimens, several of which are currently undergoing clinical testing (Table 2). Bone marrow microenvironment and stroma-mediated processes, including increased basic fibroblast growth factor (FGF2) and CXCL12/CXCR4 signaling, have been described in FLT3 inhibitor-resistant patient cases and may protect FLT3-mutated progenitors.88–92 Increased activity of several parallel prosurvival pathways also contribute to resistance. FLT3-independent activation of the RAS/RAF/MEK/ERK pathway may serve as a potential therapeutic target. 93 The PI3K/AKT/mTOR pathway is also upregulated in FLT3 TKI-resistant cases, suggesting that AKT or mTOR inhibitors may be useful in this setting.12,94–96 Constant signaling through FLT3 leads to downstream activation of STAT5 and the oncogenic serine/threonine kinase Pim-1, leading to a positive feedback loop that consolidates aberrant FLT3 signaling.97,98 A Pim kinase inhibitor is currently being studied in an early phase trial in patients with relapsed/refractory AML (ClinicalTrials.gov identifier: NCT02078609) and there is also interest in the use of STAT5 inhibitors in patients harboring FLT3 mutations.97,99,100 CDK4 and CDK6 regulate transcription of both FLT3 and Pim-1, and therefore inhibition of these kinases may target multiple pathogeneic pathways that are important in FLT3-mutated AML. 101 An ongoing trial combining sorafenib with the CDK4/6 inhibition is currently ongoing and seeks to test this hypothesis. Additionally, upregulation of anti-apoptotic proteins (e.g. Bcl-2, Bcl-xL and Mcl-1) is often observed in TKI-resistant cases and may contribute to a resistant phenotype.102–105 Notably, this process may in part be driven by aberrant Pim-1 kinase activity.106–108 Supporting these apoptotic pathways as a potential therapeutic target in FLT3-mutated AML, in vitro synergy between FLT3 and Bcl-2 inhibitors has been reported. 104 Clinical trials combining second-generation FLT3 inhibitors with venetoclax are currently planned (ClinicalTrials.gov identifier: NCT03625505).
Ongoing studies of FLT3-inhibitor-based combinations 1 .

Conventional chemotherapy and hypomethylating agent-based combinations are excluded.
Future directions
The development of FLT3 inhibitors has revolutionized the standard of care for FLT3-mutated AML, and has improved the outcomes of these patients both in the frontline and relapsed/refractory settings.41,114 With the rapid development of new, active second-generation FLT3 inhibitors with varying properties, important questions remain. In the frontline setting, the addition of the multikinase FLT3 inhibitor midostaurin to induction therapy has been shown to improve OS in younger patients with FLT3-mutated (ITD and D835) AML; however, whether better or worse results would be obtained with more specific, second-generation FLT3 TKIs such as quizartinib, gilteritinib, or crenolanib is currently unknown. Theoretically, these more specific FLT3 inhibitors may play a larger role in the relapsed/refractory setting when the disease is more monoclonal and dependent on FLT3 signaling for survival. 53 This hypothesis is being explored in ongoing randomized studies of frontline induction with second-generation FLT3 TKIs (including quizartinib, crenolanib, and gilteritinib). It is also unclear how the widespread use of FLT3 inhibitors in the frontline setting will impact the need for HSCT in first remission for these patients. While in the RATIFY study the best outcomes were observed in patients who received midostaurin with induction and then underwent subsequent HSCT in remission, it is possible that longer FLT3 inhibitor maintenance beyond 1 year or the use of alternative FLT3 inhibitors may alter our current risk stratification of patients with FLT3-mutated AML, particularly when other established prognostic factors are considered (e.g. NPM1 status, type of FLT3 mutation, and FLT3 allelic ratio). However, at this time, the recommended approach in newly diagnosed FLT3-mutated AML in fit patients remains the addition of midostaurin to intensive chemotherapy, with a plan to go to HSCT in first remission.
For those patients who do undergo HSCT in first remission, the role of FLT3 inhibitors as maintenance therapy is unclear. Studies of the first-generation TKIs sorafenib and midostaurin have suggested that they may be effective and associated with improved post-HSCT EFS and OS but may be associated with more toxicities and require frequent and liberal dose reduction in this setting.32,115–118 In contrast, preliminary evidence suggests that quizartinib, a more specific FLT3 inhibitor, is well tolerated and effective as post-HSCT maintenance. 119 A randomized study of gilteritinib versus placebo administered after HSCT in FLT3-mutated AML is ongoing and may help to more definitively address the benefit of FLT3 inhibition in this setting (BMT CTN 1506; ClinicalTrials.gov identifier: NCT02997202).
Many investigators have evaluated FLT3 inhibitors with standard chemotherapy or hypomethyla-ting agents, both of which commonly serve as backbone regimens for the study of new AML drugs. However, with an increased understanding of the intra- and extracellular mechanisms responsible for TKI resistance, rationally designed combinations with agents targeting specific resistance pathways will hopefully lead to further improved outcomes. For example, a hypomethylating agent plus the Bcl-2 inhibitor venetoclax has shown very promising activity in AML and is emerging as a potential new standard of care for the frontline treatment of older adults with newly diagnosed AML not candidates for intensive chemotherapy; 120 however, despite the preclinical rationale for Bcl-2 inhibition in FLT3-mutated AML, the role of venetoclax in this setting remains largely unknown and needs to be evaluated in rationally designed combinatorial trials. While combination studies with FLT3 inhibitors and other targeted agents such as venetoclax will initially be performed predominantly in patients with relapsed/refractory disease, promising data in this setting may ultimately support the evaluation of these novel combinations in the frontline setting possibly with the addition of a hypomethylating agent backbone.
Finally, the role of FLT3 inhibitors in patients with AML harboring wild type FLT3 remains an open question. The SORAML study showed an EFS benefit and a trend to OS benefit with longer follow up with the addition of sorafenib to standard chemotherapy in younger patients with newly diagnosed AML irrespective of FLT3 status; however, as sorafenib targets multiple kinases, it is not clear what specific properties of sorafenib were the primary drivers for these outcomes.44,65 A similar randomized phase III study of a different multikinase inhibitor, midostaurin in combination with induction chemotherapy in patients with FLT3 wildtype AML is currently ongoing, and may provide further support for the use of FLT3 inhibitors in the frontline management of patients without FLT3 mutations (ClinicalTrials.gov identifier: NCT03512197).
Footnotes
Funding
This study was supported by the MD Anderson Cancer Center Support Grant CA016672, the MD Anderson Cancer Center Leukemia SPORE CA100632, the Dick Clark Leukemia Research Fund, and the MD Anderson Moon Shots Program.
Conflict of interest statement
ND has received research funding from Daiichi-Sankyo, Novartis, Astellas and served as an advisor/consultant to Daiichi-Sankyo, Novartis, Astellas, and Arog pharmaceuticals. HK has served as an advisor/consultant to Daiichi-Sankyo. FR has served on an advisor board and received honoraria from Astellas. NS has no relevant conflicts of interest to disclose.